Tutorial 4: Large-scale Brain Atlas Analysis
This tutorial demonstrates spatial analysis of a large-scale MERFISH whole-brain atlas (~8.4 million cells across 239 brain sections from 4 animals). We use spatial_query_multi to discover spatial motifs surrounding glutamatergic neurons, examine their regional distribution, perform cross-cell gene-gene covariation analysis, and compare motif-specific covarying gene programs across brain regions.
Dataset: Allen Institute WB-MERFISH adult mouse brain atlas API class: spatial_query_multi
1. Setup
[1]:
import warnings
warnings.filterwarnings("ignore")
import os
import anndata as ad
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import seaborn as sns
from SpatialQuery import spatial_query_multi
2. Load Data
The MERFISH atlas consists of 4 animals (2 coronal, 2 sagittal) with 239 brain sections total.
[3]:
DATA_DIR = "../data/merfish_mouse_brain"
files = [f for f in os.listdir(DATA_DIR) if f.endswith(".h5ad")]
adatas = [ad.read_h5ad(os.path.join(DATA_DIR, f)) for f in files]
adatas[0]
[3]:
AnnData object with n_obs × n_vars = 1915592 × 1120
obs: 'donor_id', 'development_stage_ontology_term_id', 'sex_ontology_term_id', 'self_reported_ethnicity_ontology_term_id', 'disease_ontology_term_id', 'tissue_ontology_term_id', 'cell_type_ontology_term_id', 'assay_ontology_term_id', 'suspension_type', 'cluster_id_transfer', 'subclass_transfer', 'cluster_confidence_score', 'subclass_confidence_score', 'high_quality_transfer', 'major_brain_region', 'ccf_region_name', 'brain_section_label', 'tissue_type', 'is_primary_data', 'cell_type', 'assay', 'disease', 'sex', 'tissue', 'self_reported_ethnicity', 'development_stage', 'observation_joinid'
var: 'gene_name', 'feature_is_filtered', 'feature_name', 'feature_reference', 'feature_biotype', 'feature_length', 'feature_type'
uns: 'citation', 'organism', 'organism_ontology_term_id', 'schema_reference', 'schema_version', 'title'
obsm: 'X_CCF', 'X_spatial_coords', 'X_umap'
[4]:
# Split each animal into individual brain sections
adatas_fov = []
dataset_names = []
for adata in adatas:
dataset_name = adata.uns["title"]
for label in adata.obs["brain_section_label"].unique():
adatas_fov.append(adata[adata.obs["brain_section_label"] == label].copy())
dataset_names.extend([dataset_name] * len(adata.obs["brain_section_label"].unique()))
print(f"Total FOVs: {len(adatas_fov)}")
print(f"Total cells: {sum(a.n_obs for a in adatas_fov):,}")
Total FOVs: 239
Total cells: 8,380,288
[5]:
print(f'Max value: {adata.X.max()}, min value: {adata.X.min()}')
Max value: 6.908754825592041, min value: 0.0
Data is already log-normalized. We skip normalization during initialization (if_lognorm=False).
3. Initialize SpatialQuery Multi
[6]:
# ---- Configuration: adjust these to match your dataset ----
spatial_key = "X_spatial_coords"
label_key = "cell_type"
feature_name = "gene_name"
[7]:
spm = spatial_query_multi(
adatas_fov,
datasets=dataset_names,
spatial_key=spatial_key,
label_key=label_key,
feature_name=feature_name,
build_gene_index=False,
if_lognorm=False,
if_normalize_spatial_coord=True,
)
Replacing _ with hyphen in WB_MERFISH_animal2_coronal.
Replacing _ with hyphen in WB_MERFISH_animal2_coronal.
Replacing _ with hyphen in WB_MERFISH_animal2_coronal.
Replacing _ with hyphen in WB_MERFISH_animal2_coronal.
Replacing _ with hyphen in WB_MERFISH_animal2_coronal.
Replacing _ with hyphen in WB_MERFISH_animal2_coronal.
Replacing _ with hyphen in WB_MERFISH_animal2_coronal.
Replacing _ with hyphen in WB_MERFISH_animal2_coronal.
Replacing _ with hyphen in WB_MERFISH_animal2_coronal.
Replacing _ with hyphen in WB_MERFISH_animal2_coronal.
... (1183 lines omitted) ...
Scale factor: 0.0843
build_gene_index is False. Using adata.X for gene expression analysis.
[8]:
valid_ds_names = sorted(set(d.split("_")[0] for d in spm.datasets))
print(valid_ds_names)
['WB-MERFISH-animal1-coronal', 'WB-MERFISH-animal2-coronal', 'WB-MERFISH-animal3-sagittal', 'WB-MERFISH-animal4-sagittal']
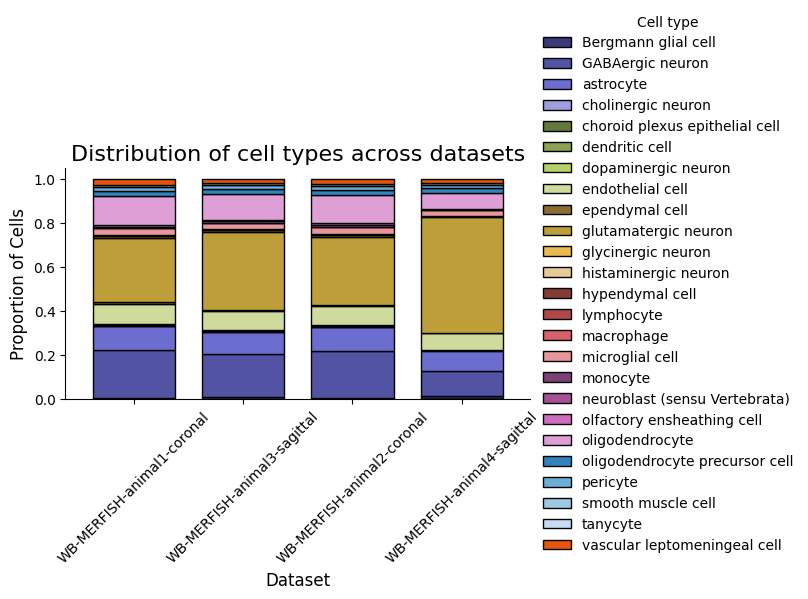
4. Cell Type Composition
[9]:
from matplotlib.colors import ListedColormap
import matplotlib.cm as cm
# Combine tab20b + tab20c for 40 unique colors (needed when >20 cell types)
all_colors = []
for name in ["tab20b", "tab20c"]:
cmap = cm.get_cmap(name)
all_colors.extend([cmap(i) for i in range(cmap.N)])
seen = set()
unique_colors = []
for c in all_colors:
key = tuple(round(v, 4) for v in c)
if key not in seen:
seen.add(key)
unique_colors.append(c)
custom_cmap = ListedColormap(unique_colors, name="tab40")
spm.plot_cell_type_distribution(colormap=custom_cmap)
/var/folders/wl/y90xsxr94l78931lqz6nyvz80000gp/T/ipykernel_31656/3661813038.py:7: MatplotlibDeprecationWarning: The get_cmap function was deprecated in Matplotlib 3.7 and will be removed in 3.11. Use ``matplotlib.colormaps[name]`` or ``matplotlib.colormaps.get_cmap()`` or ``pyplot.get_cmap()`` instead.
cmap = cm.get_cmap(name)
/var/folders/wl/y90xsxr94l78931lqz6nyvz80000gp/T/ipykernel_31656/3661813038.py:7: MatplotlibDeprecationWarning: The get_cmap function was deprecated in Matplotlib 3.7 and will be removed in 3.11. Use ``matplotlib.colormaps[name]`` or ``matplotlib.colormaps.get_cmap()`` or ``pyplot.get_cmap()`` instead.
cmap = cm.get_cmap(name)
/Users/sa3520/BWH/spatial query/python/SpatialQuery/spatial_query_multiple_fov.py:1441: MatplotlibDeprecationWarning: The get_cmap function was deprecated in Matplotlib 3.7 and will be removed in 3.11. Use ``matplotlib.colormaps[name]`` or ``matplotlib.colormaps.get_cmap()`` or ``pyplot.get_cmap()`` instead.
cmap = cm.get_cmap(colormap)
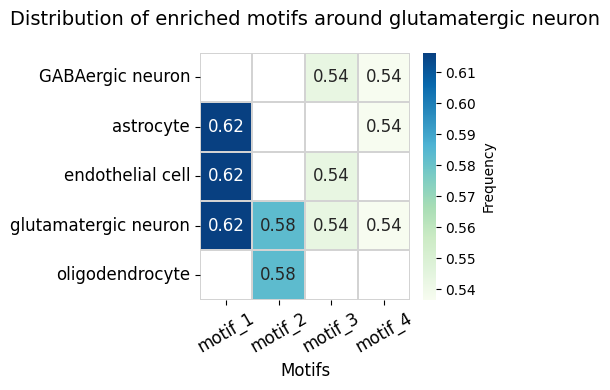
5. Motif Enrichment Analysis
Discover enriched motifs surrounding glutamatergic neurons in coronal sections. We restrict to coronal datasets for consistency across animals.
[10]:
anchor_ct = 'glutamatergic neuron'
max_dist = 5
min_support = 0.5
coronal_ds = ['WB-MERFISH-animal1-coronal', 'WB-MERFISH-animal2-coronal']
motif_glut_coronal_df, motif_glut_coronal_motif_id, motif_glut_coronal_center_id = spm.motif_enrichment_dist(
ct=anchor_ct,
dataset=coronal_ds,
max_dist=max_dist,
min_support=min_support,
return_cellID=True
)
[11]:
from SpatialQuery.plotting import plot_motif_enrichment_heatmap
plot_motif_enrichment_heatmap(enrich_df=motif_glut_coronal_df,
figsize=(5, 4),
)
[12]:
# Collect motif+ / motif- obs for each motif
data_by_motif = {}
for motif_idx in range(len(motif_glut_coronal_df)):
motifs_list = motif_glut_coronal_df.iloc[motif_idx]['motifs']
outer_key = str(sorted(motifs_list))
center_id_dict = motif_glut_coronal_center_id.get(outer_key, {})
pos_obs_list, neg_obs_list = [], []
for sp, fov_ds in zip(spm.spatial_queries, spm.datasets):
if not any(fov_ds.startswith(td) for td in coronal_ds):
continue
obs = sp.adata.obs.copy()
ct_mask = obs[label_key] == anchor_ct
pos_indices = center_id_dict.get(fov_ds, [])
if len(pos_indices) > 0:
pos_mask = np.zeros(len(obs), dtype=bool)
pos_mask[np.asarray(pos_indices)] = True
pos_obs_list.append(obs[pos_mask & ct_mask])
neg_obs_list.append(obs[ct_mask & ~pos_mask])
else:
neg_obs_list.append(obs[ct_mask])
pos_obs = pd.concat(pos_obs_list, ignore_index=True) if pos_obs_list else pd.DataFrame()
neg_obs = pd.concat(neg_obs_list, ignore_index=True) if neg_obs_list else pd.DataFrame()
motif_label = '+'.join(sorted([m for m in motifs_list if m != anchor_ct])) or 'Self'
data_by_motif[motif_idx] = dict(pos_obs=pos_obs, neg_obs=neg_obs, motif_label=motif_label)
[13]:
# Subclass composition (motif+ vs motif-) per motif
import matplotlib.patches as mpatches
all_obs = pd.concat(
[d['pos_obs'] for d in data_by_motif.values()] +
[d['neg_obs'] for d in data_by_motif.values()],
ignore_index=True
)
top_subs = all_obs['subclass_transfer'].value_counts().index[:8].tolist()
n_motifs = len(data_by_motif)
bar_w, gap = 0.35, 0.05
x_centers = np.arange(n_motifs) * 1.3
cmap = plt.cm.get_cmap('Set2', len(top_subs))
sub_colors = {s: cmap(i) for i, s in enumerate(top_subs)}
fig, ax = plt.subplots(figsize=(max(6, n_motifs * 2), 5))
for motif_idx, d in data_by_motif.items():
for obs_df, x_bar in [(d['pos_obs'], x_centers[motif_idx]),
(d['neg_obs'], x_centers[motif_idx] + bar_w + gap)]:
if len(obs_df) == 0:
continue
counts = obs_df['subclass_transfer'].value_counts(normalize=True)
top_fracs = {sub: counts.get(sub, 0.0) for sub in top_subs}
total_top = sum(top_fracs.values())
if total_top == 0:
continue
bottom = 0.0
for sub in top_subs:
frac = top_fracs[sub] / total_top
ax.bar(x_bar, frac, bar_w, bottom=bottom, color=sub_colors[sub])
bottom += frac
tick_pos = [x_centers[i] + (bar_w + gap) / 2 for i in range(n_motifs)]
ax.set_xticks(tick_pos)
ax.set_xticklabels([f'Motif-{i+1}' for i in range(n_motifs)])
for i in range(n_motifs):
ax.text(x_centers[i], -0.05, '+', ha='center', va='top', fontsize=7, transform=ax.get_xaxis_transform())
ax.text(x_centers[i] + bar_w + gap, -0.05, '−', ha='center', va='top', fontsize=7, transform=ax.get_xaxis_transform())
ax.legend(handles=[mpatches.Patch(color=sub_colors[s], label=s) for s in top_subs],
loc='center left', bbox_to_anchor=(1, 0.5), frameon=False)
ax.set_ylabel('Fraction')
ax.set_title('Subclass composition (Motif+ vs Motif−)')
plt.tight_layout()
/var/folders/wl/y90xsxr94l78931lqz6nyvz80000gp/T/ipykernel_31656/514646767.py:14: MatplotlibDeprecationWarning: The get_cmap function was deprecated in Matplotlib 3.7 and will be removed in 3.11. Use ``matplotlib.colormaps[name]`` or ``matplotlib.colormaps.get_cmap()`` or ``pyplot.get_cmap()`` instead.
cmap = plt.cm.get_cmap('Set2', len(top_subs))
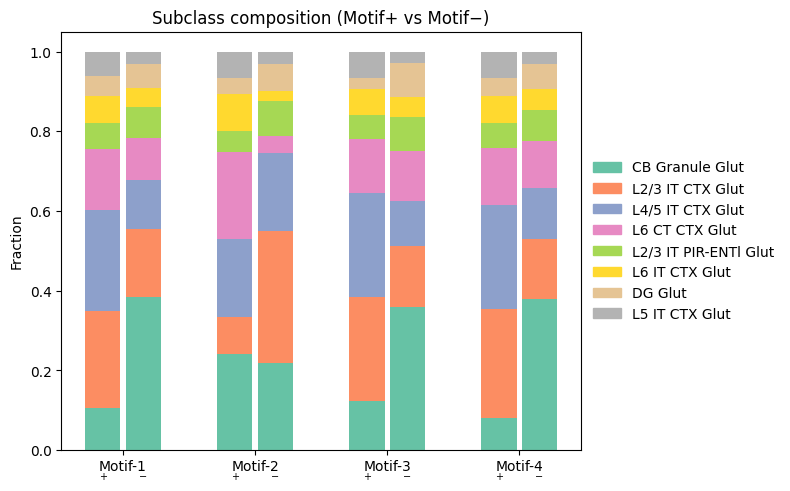
[14]:
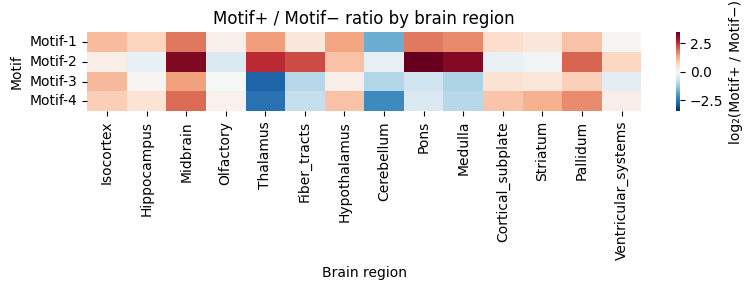
# Heatmap: Motif+ / Motif- ratio per brain region
region_order = list(data_by_motif[0]['pos_obs']['major_brain_region'].value_counts().index)
rows = {}
for motif_idx, d in data_by_motif.items():
pos_region = d['pos_obs']['major_brain_region'].value_counts()
neg_region = d['neg_obs']['major_brain_region'].value_counts()
ratio = pd.Series({
r: pos_region.get(r, 0) / max(neg_region.get(r, 0), 1)
for r in region_order
})
rows[f'Motif-{motif_idx+1}'] = ratio
hm_df = pd.DataFrame(rows, index=region_order).T
log2_df = np.log2(hm_df.clip(lower=1e-6))
abs_max = np.nanmax(np.abs(log2_df.values))
fig, ax = plt.subplots(figsize=(8, max(3, n_motifs * 0.6)))
sns.heatmap(
log2_df, ax=ax, cmap='RdBu_r',
vmin=-abs_max, vmax=abs_max, center=0,
cbar_kws={'label': 'log₂(Motif+ / Motif−)'},
)
ax.set_xticklabels(ax.get_xticklabels(), rotation=90)
ax.set_yticklabels(ax.get_yticklabels(), rotation=0)
ax.set_xlabel('Brain region')
ax.set_ylabel('Motif')
ax.set_title('Motif+ / Motif− ratio by brain region')
plt.tight_layout()
[15]:
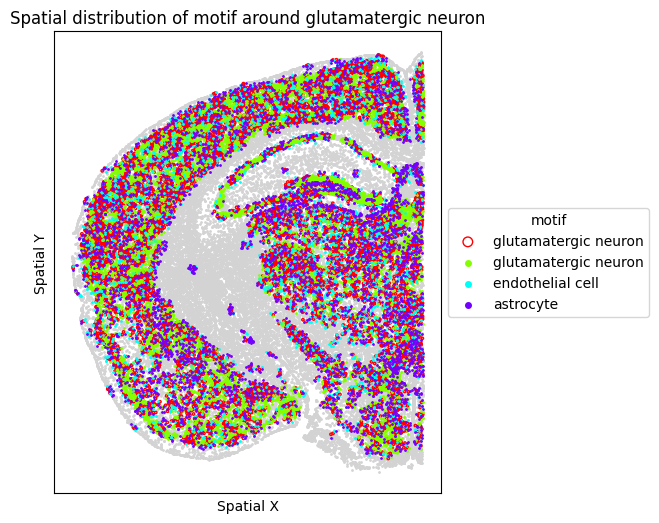
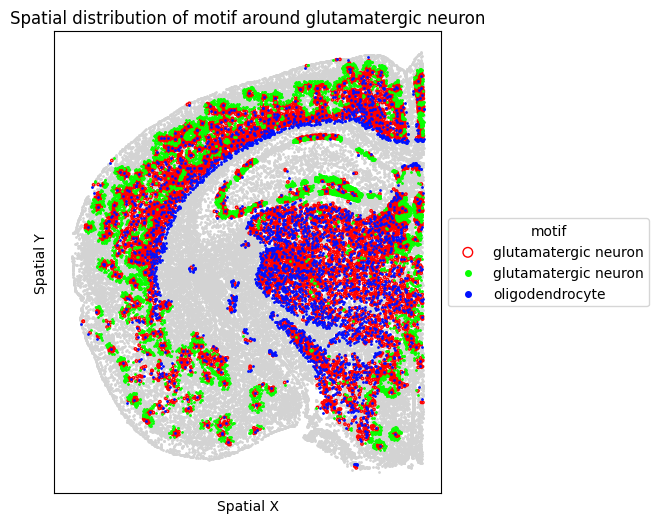
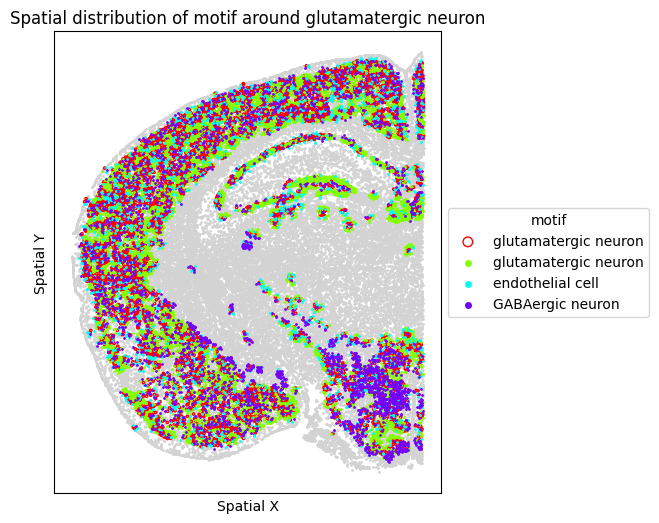
# Visualize motif distribution in a single representative FOV
target_fov = "WB-MERFISH-animal2-coronal_49"
target_sp = [sp for sp in spm.spatial_queries if sp.dataset == target_fov][0]
for m in motif_glut_coronal_df["motifs"]:
target_sp.plot_motif_celltype(
ct=anchor_ct,
motif=m,
max_dist=max_dist,
figsize=(5, 6),
)
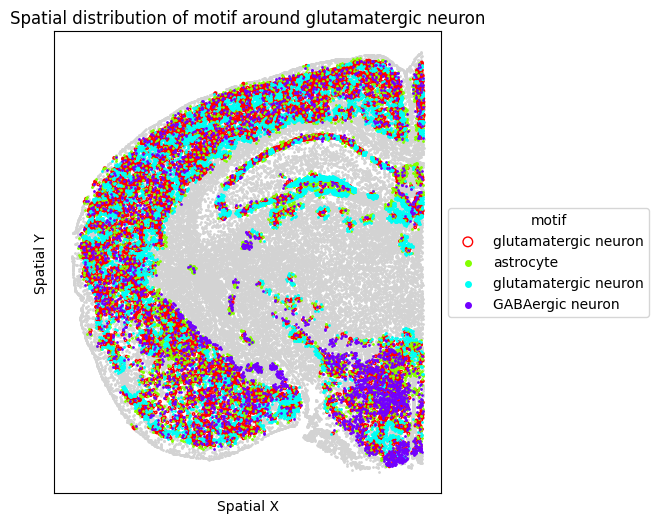
6. Cross-cell Gene-Gene Covariation
Identify gene pairs with spatially-specific cross-cell correlation between glutamatergic neurons and their motif neighbors.
[16]:
print(f"Anchor: {anchor_ct}, max_dist: {max_dist}")
print(f"Coronal datasets: {coronal_ds}")
Anchor: glutamatergic neuron, max_dist: 5
Coronal datasets: ['WB-MERFISH-animal1-coronal', 'WB-MERFISH-animal2-coronal']
[17]:
motif2 = ['oligodendrocyte', 'glutamatergic neuron']
covarying_genes_m2 = spm.compute_gene_gene_correlation_by_type(
ct=anchor_ct,
motif=motif2,
dataset=coronal_ds,
max_dist=max_dist,
)
Computing covarying genes using expression data ...
Only one non-center cell type in motif: ['oligodendrocyte']. Using compute_gene_gene_correlation method.
No genes specified. Using union of genes across all selected FOVs ...
Gene coverage: 1120 genes in all FOVs, 1120 genes total (union)
Analyzing 1120 genes across 213 FOVs
================================================================================
Step 1: Computing and accumulating statistics across FOVs
================================================================================
... (1523 lines omitted) ...
Total gene pairs analyzed: 1254400
Significant pairs: 151030
[18]:
covarying_genes_m2 = covarying_genes_m2[covarying_genes_m2['if_significant']]
covarying_genes_m2
[18]:
| gene_center | gene_motif | corr_neighbor | corr_non_neighbor | p_value_test1 | delta_corr_test1 | corr_center_no_motif | p_value_test2 | delta_corr_test2 | combined_score | adj-pval-test1 | adj-pval-test2 | if_significant | cell_type | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | Prkcd | Prkcd | 0.483658 | -0.025477 | 0.0 | 0.509135 | 0.066700 | 0.0 | 0.416958 | 133.383376 | 0.0 | 0.0 | True | oligodendrocyte |
| 1 | Grik3 | Slc17a7 | 0.200625 | -0.046645 | 0.0 | 0.247271 | -0.169207 | 0.0 | 0.369833 | 99.919199 | 0.0 | 0.0 | True | oligodendrocyte |
| 2 | Tnnt1 | Prkcd | 0.318601 | -0.015084 | 0.0 | 0.333685 | 0.008595 | 0.0 | 0.310006 | 95.132929 | 0.0 | 0.0 | True | oligodendrocyte |
| 3 | Grid2ip | Prkcd | 0.353341 | -0.021876 | 0.0 | 0.375217 | 0.065240 | 0.0 | 0.288100 | 94.270543 | 0.0 | 0.0 | True | oligodendrocyte |
| 4 | Rgs16 | Prkcd | 0.316480 | -0.020070 | 0.0 | 0.336550 | 0.023593 | 0.0 | 0.292886 | 91.795601 | 0.0 | 0.0 | True | oligodendrocyte |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 1254395 | Gucy1a1 | Slc17a7 | -0.098372 | 0.052502 | 0.0 | -0.150874 | 0.172281 | 0.0 | -0.270654 | -70.415942 | 0.0 | 0.0 | True | oligodendrocyte |
| 1254396 | Cux2 | Slc17a7 | -0.146541 | 0.030721 | 0.0 | -0.177261 | 0.150265 | 0.0 | -0.296806 | -78.282743 | 0.0 | 0.0 | True | oligodendrocyte |
| 1254397 | Otof | Slc17a7 | -0.153165 | 0.065437 | 0.0 | -0.218603 | 0.149220 | 0.0 | -0.302385 | -83.175139 | 0.0 | 0.0 | True | oligodendrocyte |
| 1254398 | Calb1 | Slc17a7 | -0.166215 | 0.052068 | 0.0 | -0.218283 | 0.136621 | 0.0 | -0.302836 | -83.241135 | 0.0 | 0.0 | True | oligodendrocyte |
| 1254399 | Syt17 | Slc17a7 | -0.115302 | 0.056794 | 0.0 | -0.172096 | 0.233440 | 0.0 | -0.348742 | -88.724367 | 0.0 | 0.0 | True | oligodendrocyte |
151030 rows × 14 columns
[19]:
# Covarying gene pairs for motif2 (oligodendrocyte)
oligo_pos = covarying_genes_m2[covarying_genes_m2['cell_type'] == 'oligodendrocyte'].copy()
oligo_pos = oligo_pos[oligo_pos['combined_score'] > 0]
top_gene_center = pd.DataFrame(oligo_pos['gene_center'].value_counts().head(20))[::-1]
top_gene_motif = pd.DataFrame(oligo_pos['gene_motif'].value_counts().head(20))[::-1]
fig, axs = plt.subplots(1, 3, figsize=(12, 5))
axs[0].barh(top_gene_center.index.tolist(), top_gene_center['count'].tolist(), color='salmon')
axs[0].set_title(f'{anchor_ct} genes')
axs[1].barh(top_gene_motif.index.tolist(), top_gene_motif['count'].tolist(), color='skyblue')
axs[1].set_title('Oligodendrocyte genes')
# Heatmap of top50 gene pairs
from scipy.cluster.hierarchy import linkage, dendrogram
from scipy.spatial.distance import pdist
top20_center = oligo_pos['gene_center'].value_counts().head(20).index.tolist()
top20_motif = oligo_pos['gene_motif'].value_counts().head(20).index.tolist()
sub = oligo_pos[oligo_pos['gene_center'].isin(top20_center) & oligo_pos['gene_motif'].isin(top20_motif)]
pivot = sub.pivot_table(index='gene_center', columns='gene_motif', values='combined_score', fill_value=0)
mask = (pivot == 0)
data = np.log10(pivot + 1)
row_order = dendrogram(linkage(pdist(data), method='average'), no_plot=True)['leaves']
col_order = dendrogram(linkage(pdist(data.T), method='average'), no_plot=True)['leaves']
sns.heatmap(
data.iloc[row_order, col_order],
mask=mask.iloc[row_order, col_order],
cmap='Reds',
cbar_kws={'label': 'log10(score + 1)'},
ax=axs[2],
)
plt.tight_layout()
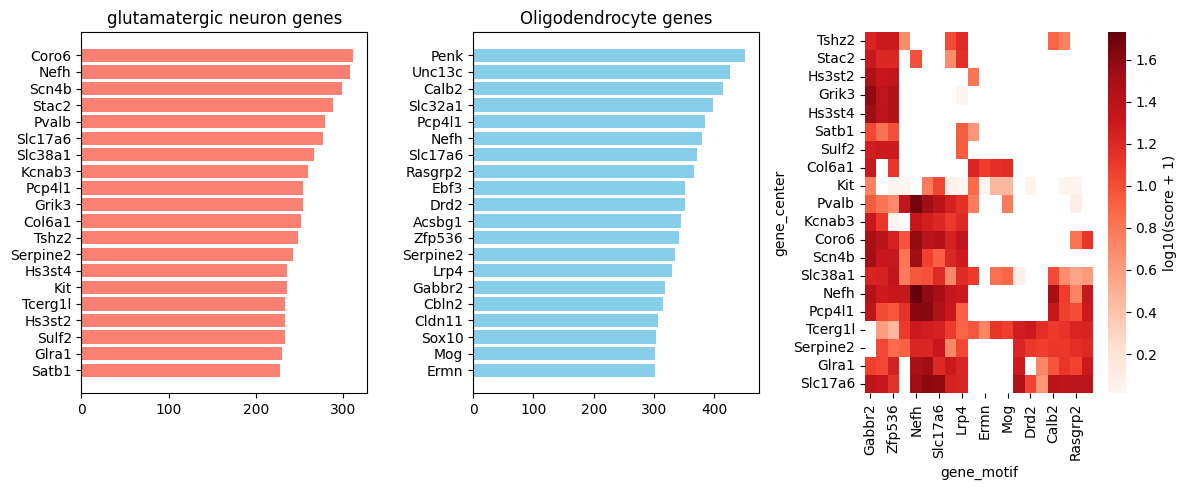
7. Regional Stratification: Isocortex Analysis
Since glutamatergic neurons are most abundant in the Isocortex, we construct a subset containing only Isocortex cells and repeat the covariation analysis to examine whether motif-specific gene programs are preserved in a single brain region.
[20]:
# Subset to Isocortex cells from coronal sections
adatas_coronal_isocortex = []
dataset_names_coronal_isocortex = []
adatas_coronal = [adatas_fov[i] for i in range(len(adatas_fov)) if "coronal" in adatas_fov[i].uns["title"]]
for adata in adatas_coronal:
sub_data = adata[adata.obs["major_brain_region"] == "Isocortex"].copy()
adatas_coronal_isocortex.append(sub_data)
dataset_names_coronal_isocortex.append(adata.uns["title"])
print(f"Isocortex FOVs: {len(adatas_coronal_isocortex)}")
print(f"Isocortex cells: {sum(a.n_obs for a in adatas_coronal_isocortex):,}")
Isocortex FOVs: 213
Isocortex cells: 1,337,180
[21]:
spm_coronal_isocortex = spatial_query_multi(
adatas=adatas_coronal_isocortex,
datasets=dataset_names_coronal_isocortex,
spatial_key=spatial_key,
label_key=label_key,
build_gene_index=False,
feature_name=feature_name,
if_lognorm=False,
if_normalize_spatial_coord=True
)
Replacing _ with hyphen in WB_MERFISH_animal2_coronal.
Replacing _ with hyphen in WB_MERFISH_animal2_coronal.
Replacing _ with hyphen in WB_MERFISH_animal2_coronal.
Replacing _ with hyphen in WB_MERFISH_animal2_coronal.
Replacing _ with hyphen in WB_MERFISH_animal2_coronal.
Replacing _ with hyphen in WB_MERFISH_animal2_coronal.
Replacing _ with hyphen in WB_MERFISH_animal2_coronal.
Replacing _ with hyphen in WB_MERFISH_animal2_coronal.
Replacing _ with hyphen in WB_MERFISH_animal2_coronal.
Replacing _ with hyphen in WB_MERFISH_animal2_coronal.
... (1053 lines omitted) ...
Scale factor: 0.0881
build_gene_index is False. Using adata.X for gene expression analysis.
[22]:
motif0 = motif_glut_coronal_df['motifs'].iloc[0]
covarying_isocortex_motif0 = spm_coronal_isocortex.compute_gene_gene_correlation_by_type(
ct=anchor_ct,
motif=motif0,
dataset=coronal_ds,
max_dist=max_dist,
)
Computing covarying genes using expression data ...
Analyzing 2 non-center cell types in motif: ['astrocyte', 'endothelial cell']
================================================================================
Selected 213 FOVs for analysis
No genes specified. Using union of genes across all selected FOVs ...
Gene coverage: 1120 genes in all FOVs, 1120 genes total (union)
Analyzing 1120 genes across 213 FOVs
================================================================================
Step 1: Computing Correlation-3 (Center without motif vs Neighbors)
... (938 lines omitted) ...
Results prepared and sorted
[23]:
motif3 = motif_glut_coronal_df['motifs'].iloc[3]
covarying_isocortex_motif3 = spm_coronal_isocortex.compute_gene_gene_correlation_by_type(
ct=anchor_ct,
motif=motif3,
dataset=coronal_ds,
max_dist=max_dist,
)
Computing covarying genes using expression data ...
Analyzing 2 non-center cell types in motif: ['GABAergic neuron', 'astrocyte']
================================================================================
Selected 213 FOVs for analysis
No genes specified. Using union of genes across all selected FOVs ...
Gene coverage: 1120 genes in all FOVs, 1120 genes total (union)
Analyzing 1120 genes across 213 FOVs
================================================================================
Step 1: Computing Correlation-3 (Center without motif vs Neighbors)
... (938 lines omitted) ...
Results prepared and sorted
[24]:
covarying_isocortex_motif3_astro = covarying_isocortex_motif3[covarying_isocortex_motif3['if_significant']]
covarying_isocortex_motif3_astro = covarying_isocortex_motif3_astro[covarying_isocortex_motif3_astro['cell_type'] == 'astrocyte']
covarying_isocortex_motif0_astro = covarying_isocortex_motif0[covarying_isocortex_motif0['if_significant']]
covarying_isocortex_motif0_astro = covarying_isocortex_motif0_astro[covarying_isocortex_motif0_astro['cell_type'] == 'astrocyte']
[25]:
rank_genes_glut_astro_isocortex = spm_coronal_isocortex.test_score_difference(
result_A=covarying_isocortex_motif0_astro,
result_B=covarying_isocortex_motif3_astro,
percentile_threshold=99
)
Significant pairs in A: 6652
Significant pairs in B: 7929
Pairs that are significant in at least one group: 9350
Time to compute percentiles: 0.11 seconds.
============================================================
Score Difference Test Results
============================================================
Total pairs tested: 5231
Outlier pairs (percentile > 99 or < 1): 105
... (4 lines omitted) ...
Mean score difference: 0.199
Std score difference: 5.155
[26]:
df_plot = rank_genes_glut_astro_isocortex[rank_genes_glut_astro_isocortex['outlier_direction'] != 'not_outlier'].copy()
m0 = df_plot[df_plot['outlier_direction'] == 'higher_in_A'].nlargest(100, 'score_diff')
m3 = df_plot[df_plot['outlier_direction'] == 'lower_in_A'].nsmallest(100, 'score_diff')
df_plot = pd.concat([m0, m3])
center_shared = sorted(set(m0['gene_center']) & set(m3['gene_center']))
center_m0_only = sorted(set(m0['gene_center']) - set(m3['gene_center']))
center_m3_only = sorted(set(m3['gene_center']) - set(m0['gene_center']))
center_genes = center_m3_only + center_shared + center_m0_only
motif_shared = sorted(set(m0['gene_motif']) & set(m3['gene_motif']))
motif_m0_only = sorted(set(m0['gene_motif']) - set(m3['gene_motif']))
motif_m3_only = sorted(set(m3['gene_motif']) - set(m0['gene_motif']))
motif_genes = motif_m3_only + motif_shared + motif_m0_only
n_row = len(center_genes)
n_col = len(motif_genes)
row_idx = {g: i for i, g in enumerate(center_genes)}
col_idx = {g: i for i, g in enumerate(motif_genes)}
mat = np.full((n_row, n_col), np.nan)
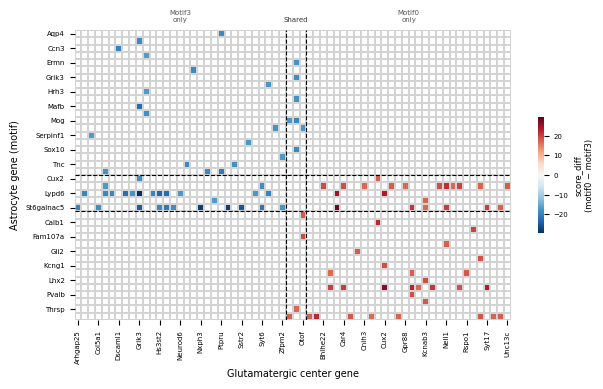
for _, row in df_plot.iterrows():
mat[row_idx[row['gene_center']], col_idx[row['gene_motif']]] = row['score_diff']
vmax = np.nanmax(np.abs(mat))
mat_df = pd.DataFrame(mat, index=center_genes, columns=motif_genes)
mat_df_T = mat_df.T # rows = astrocyte genes, cols = glut genes
fig, ax = plt.subplots(figsize=(max(4, n_row * 0.1), max(3, n_col * 0.1)))
sns.heatmap(
mat_df_T,
cmap='RdBu_r',
center=0,
vmin=-vmax, vmax=vmax,
linewidths=0.01,
linecolor='lightgrey',
mask=np.isnan(mat.T),
cbar_kws={'label': 'score_diff\n(motif0 − motif3)', 'shrink': 0.4},
ax=ax,
)
cbar = ax.collections[0].colorbar
cbar.ax.tick_params(labelsize=5)
cbar.set_label('score_diff\n(motif0 − motif3)', fontsize=6)
# Dividing lines — seaborn heatmap: col x goes 0..n_col, row y goes 0..n_row
n_m3_only_col = len(motif_m3_only)
n_shared_col = len(motif_shared)
n_m3_only_row = len(center_m3_only)
n_shared_row = len(center_shared)
# After transpose: rows = motif genes, cols = center genes
# Dashed lines: column boundaries from original row splits, row boundaries from col splits
for x in [n_m3_only_row, n_m3_only_row + n_shared_row]:
ax.axvline(x, color='black', linewidth=0.8, linestyle='--')
for y in [n_m3_only_col, n_m3_only_col + n_shared_col]:
ax.axhline(y, color='black', linewidth=0.8, linestyle='--')
ax.set_xlabel('Glutamatergic center gene', fontsize=7)
ax.set_ylabel('Astrocyte gene (motif)', fontsize=7)
ax.tick_params(axis='x', labelsize=5)
ax.tick_params(axis='y', labelsize=5)
# Region annotations: columns are center genes (motif3-only | shared | motif0-only)
ax.text(n_m3_only_row / 2, -1.2, 'Motif3\nonly', ha='center', fontsize=5, color='#555')
ax.text(n_m3_only_row + n_shared_row / 2, -1.2, 'Shared', ha='center', fontsize=5, color='#333')
ax.text(n_m3_only_row + n_shared_row + len(center_m0_only) / 2, -1.2, 'Motif0\nonly', ha='center', fontsize=5, color='#555')
plt.tight_layout()
plt.show()