Tutorial 3: Spatial Proteomics Analysis of Tumor Microenvironment
This tutorial demonstrates multi-condition spatial analysis using CODEX spatial proteomics data from colorectal cancer (CRC) tissue. The dataset contains two immune subtypes — CLR (Crohn’s-Like Reaction) and DII (Diffuse Inflammatory Infiltration) — each with multiple fields of view (FOVs).
We use spatial_query_multi to compare spatial motif patterns between conditions, perform differential motif analysis, and examine hypothesis-driven motifs such as tertiary lymphoid structures (TLS) and immunosuppressive niches.
API class: spatial_query_multi
1. Setup
[1]:
import warnings
warnings.filterwarnings("ignore")
import os
import anndata as ad
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import seaborn as sns
from SpatialQuery import spatial_query_multi
2. Load & Preprocess Data
The CODEX data contains raw protein intensities. We assign disease state labels, apply z-score normalization, and split by FOV for multi-dataset analysis.
[3]:
# ---- Configuration: adjust these to match your dataset ----
DATA_DIR = "../data/codex_cancer"
adata = ad.read_h5ad(f"{DATA_DIR}/codex_data.h5ad")
adata
[3]:
AnnData object with n_obs × n_vars = 258385 × 56
obs: 'CellID', 'ClusterID', 'EventID', 'File Name', 'Region', 'TMA_AB', 'TMA_12', 'Index in File', 'groups', 'patients', 'spots', 'cell_id', 'size:size', 'HOECHST1_Cyc_1_ch_1', 'DRAQ5_Cyc_23_ch_4', 'Profile_Homogeneity:Fiter1', 'ClusterSize', 'ClusterName', 'neighborhood10', 'CD4+ICOS+', 'CD4+Ki67+', 'CD4+PD-1+', 'CD68+CD163+ICOS+', 'CD68+CD163+Ki67+', 'CD68+CD163+PD-1+', 'CD68+ICOS+', 'CD68+Ki67+', 'CD68+PD-1+', 'CD8+ICOS+', 'CD8+Ki67+', 'CD8+PD-1+', 'Treg-ICOS+', 'Treg-Ki67+', 'Treg-PD-1+', 'neighborhood number final', 'neighborhood name'
var: 'marker_name', 'full_name', 'cell_type_annotation', 'cycle', 'channel'
uns: 'data_source', 'groups_mapping', 'n_markers', 'technology'
obsm: 'X_spatial_global', 'X_spatial_tile'
[4]:
# Assign immune subtype labels based on group ID
adata.obs["state"] = "CLR"
adata.obs["state"][adata.obs["groups"] == 2] = "DII"
print(f"Max value: {adata.X.max()}")
Max value: 54776.6953125
[5]:
# Feature-wise z-score normalization for protein expression
adata.X = (adata.X - adata.X.mean(axis=0)) / adata.X.std(axis=0)
[6]:
# Split data by FOV and immune subtype
fov_key = "File Name"
clr_data = adata[adata.obs["state"] == "CLR"]
dii_data = adata[adata.obs["state"] == "DII"]
clr_datas = [clr_data[clr_data.obs[fov_key] == f] for f in clr_data.obs[fov_key].unique()]
dii_datas = [dii_data[dii_data.obs[fov_key] == f] for f in dii_data.obs[fov_key].unique()]
ds_names = ["CLR"] * len(clr_datas) + ["DII"] * len(dii_datas)
adata_fovs = clr_datas + dii_datas
3. Initialize SpatialQuery Multi
Since this is protein data is z-score normalized, we set if_lognorm=False.
[7]:
# ---- Configuration: adjust these to match your dataset ----
spatial_key = "X_spatial_tile"
label_key = "ClusterName"
feature_name = "marker_name"
[8]:
spm = spatial_query_multi(
adatas=adata_fovs,
datasets=ds_names,
spatial_key=spatial_key,
label_key=label_key,
feature_name=feature_name,
build_gene_index=False,
if_lognorm=False, # data is z-score normalized beforehand
if_normalize_spatial_coord=True,
)
Auto-normalizing spatial coordinates: mean nearest neighbor distance = 1.0
Scale factor: 0.0448
build_gene_index is False. Using adata.X for gene expression analysis.
Auto-normalizing spatial coordinates: mean nearest neighbor distance = 1.0
Scale factor: 0.0311
build_gene_index is False. Using adata.X for gene expression analysis.
Auto-normalizing spatial coordinates: mean nearest neighbor distance = 1.0
... (548 lines omitted) ...
Scale factor: 0.0421
build_gene_index is False. Using adata.X for gene expression analysis.
4. Cell Type Composition
[9]:
from matplotlib.colors import ListedColormap
import matplotlib.cm as cm
# Combine tab20b + tab20c for 40 unique colors
all_colors = []
for name in ["tab20b", "tab20c"]:
cmap = cm.get_cmap(name)
all_colors.extend([cmap(i) for i in range(cmap.N)])
seen = set()
unique_colors = []
for c in all_colors:
key = tuple(round(v, 4) for v in c)
if key not in seen:
seen.add(key)
unique_colors.append(c)
custom_cmap = ListedColormap(unique_colors, name="tab40")
spm.plot_cell_type_distribution(colormap=custom_cmap)
/var/folders/wl/y90xsxr94l78931lqz6nyvz80000gp/T/ipykernel_18164/676858651.py:7: MatplotlibDeprecationWarning: The get_cmap function was deprecated in Matplotlib 3.7 and will be removed in 3.11. Use ``matplotlib.colormaps[name]`` or ``matplotlib.colormaps.get_cmap()`` or ``pyplot.get_cmap()`` instead.
cmap = cm.get_cmap(name)
/var/folders/wl/y90xsxr94l78931lqz6nyvz80000gp/T/ipykernel_18164/676858651.py:7: MatplotlibDeprecationWarning: The get_cmap function was deprecated in Matplotlib 3.7 and will be removed in 3.11. Use ``matplotlib.colormaps[name]`` or ``matplotlib.colormaps.get_cmap()`` or ``pyplot.get_cmap()`` instead.
cmap = cm.get_cmap(name)
/Users/sa3520/BWH/spatial query/python/SpatialQuery/spatial_query_multiple_fov.py:1441: MatplotlibDeprecationWarning: The get_cmap function was deprecated in Matplotlib 3.7 and will be removed in 3.11. Use ``matplotlib.colormaps[name]`` or ``matplotlib.colormaps.get_cmap()`` or ``pyplot.get_cmap()`` instead.
cmap = cm.get_cmap(colormap)
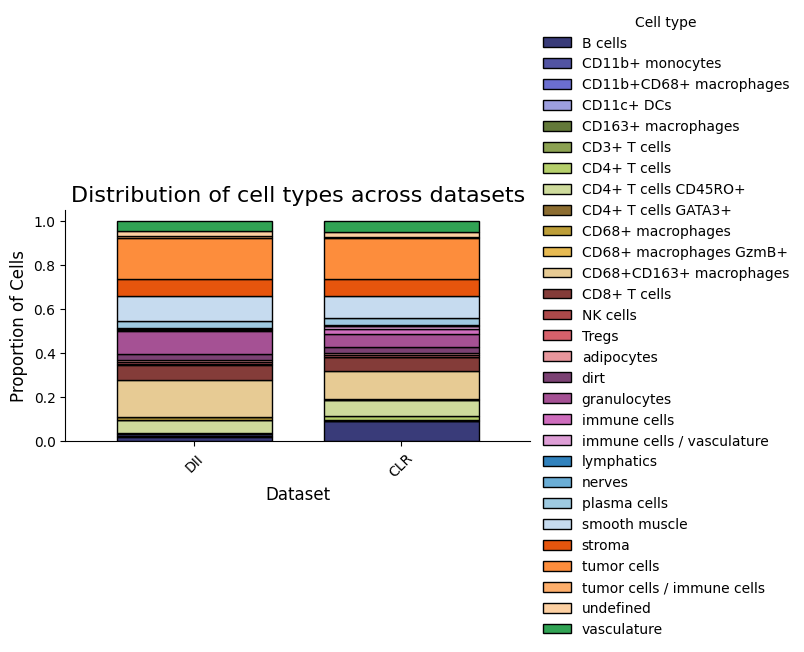
5. Motif Enrichment by Condition
Discover enriched motifs surrounding B cells in each immune subtype separately, then compare the results side by side.
[10]:
anchor_ct = 'B cells'
max_dist = 5
min_support = 0.5
clr = 'CLR'
dii = 'DII'
enrich_motif_dataset = dict()
for ds in [clr, dii]:
enrich_motif_dataset[ds] = spm.motif_enrichment_dist(
ct=anchor_ct,
dataset=ds,
max_dist=max_dist,
min_support=min_support,
return_cellID=False
)
enrich_motif_dataset[ds] = enrich_motif_dataset[ds][enrich_motif_dataset[ds]['if_significant']]
print(f"Found {len(enrich_motif_dataset[ds])} enriched motifs in {ds}")
enrich_motif_dataset_df = pd.DataFrame()
for ds, df in enrich_motif_dataset.items():
df['dataset'] = ds
enrich_motif_dataset_df = pd.concat([enrich_motif_dataset_df, df], axis=0)
Found 2 enriched motifs in CLR
Found 14 enriched motifs in DII
[11]:
enrich_motif_dataset_df
[11]:
| center | motifs | n_center_motif | n_center | n_motif | expectation | p-values | adj-pval | if_significant | dataset | |
|---|---|---|---|---|---|---|---|---|---|---|
| 0 | B cells | [B cells, CD4+ T cells CD45RO+, CD8+ T cells] | 6680 | 10269 | 27291 | 2523.399564 | 0.000000e+00 | 0.000000e+00 | True | CLR |
| 1 | B cells | [B cells, tumor cells] | 5382 | 10269 | 24255 | 2242.682805 | 0.000000e+00 | 0.000000e+00 | True | CLR |
| 0 | B cells | [B cells, CD8+ T cells] | 1704 | 2774 | 26742 | 503.531726 | 0.000000e+00 | 0.000000e+00 | True | DII |
| 1 | B cells | [B cells, CD4+ T cells CD45RO+] | 1647 | 2774 | 24170 | 455.102902 | 0.000000e+00 | 0.000000e+00 | True | DII |
| 2 | B cells | [B cells, stroma] | 1592 | 2774 | 26083 | 491.123252 | 0.000000e+00 | 0.000000e+00 | True | DII |
| 3 | B cells | [B cells, CD68+CD163+ macrophages] | 1495 | 2774 | 30610 | 576.363254 | 0.000000e+00 | 0.000000e+00 | True | DII |
| 4 | B cells | [B cells, smooth muscle] | 1425 | 2774 | 19963 | 375.888260 | 0.000000e+00 | 0.000000e+00 | True | DII |
| 5 | B cells | [CD4+ T cells CD45RO+, CD8+ T cells] | 1668 | 2774 | 58370 | 1099.063153 | 1.527325e-107 | 3.818311e-107 | True | DII |
| 6 | B cells | [CD8+ T cells, smooth muscle] | 1393 | 2774 | 47767 | 899.416646 | 1.183362e-85 | 2.535775e-85 | True | DII |
| 7 | B cells | [CD4+ T cells CD45RO+, stroma] | 1566 | 2774 | 57711 | 1086.654679 | 3.314561e-77 | 6.214802e-77 | True | DII |
| 8 | B cells | [CD8+ T cells, stroma] | 1655 | 2774 | 64677 | 1217.819215 | 7.365512e-64 | 1.227585e-63 | True | DII |
| 9 | B cells | [vasculature] | 1641 | 2774 | 66663 | 1255.214100 | 3.938264e-50 | 5.907396e-50 | True | DII |
| 10 | B cells | [CD8+ T cells, tumor cells] | 1447 | 2774 | 61444 | 1156.944259 | 1.881974e-29 | 2.566328e-29 | True | DII |
| 11 | B cells | [CD4+ T cells CD45RO+, CD68+CD163+ macrophages] | 1557 | 2774 | 70499 | 1327.443091 | 5.381246e-19 | 6.726557e-19 | True | DII |
| 12 | B cells | [CD68+CD163+ macrophages, CD8+ T cells] | 1709 | 2774 | 82157 | 1546.954454 | 1.412683e-10 | 1.630019e-10 | True | DII |
| 13 | B cells | [stroma, tumor cells] | 1419 | 2774 | 69652 | 1311.494719 | 1.712935e-05 | 1.835287e-05 | True | DII |
[12]:
enrich = enrich_motif_dataset_df.copy()
enrich["frequency"] = enrich["n_center_motif"] / enrich["n_center"]
# Sort by dataset, then by frequency within each dataset
enrich = enrich.sort_values(by=["dataset", "frequency"], ascending=[True, False]).reset_index(drop=True)
# Assign motif labels per dataset
enrich["motif_num"] = enrich.groupby("dataset").cumcount() + 1
enrich["motif_group"] = enrich["dataset"] + "_motif_" + enrich["motif_num"].astype(str)
# Build heatmap: cell types × motif groups
enrich_expanded = enrich.explode("motifs")
heatmap_data = enrich_expanded.pivot_table(
index="motifs", columns="motif_group", values="frequency", aggfunc="first"
)
col_order = enrich["motif_group"].tolist()
heatmap_data = heatmap_data[col_order]
heatmap_data.columns = [col.replace("_", "-") for col in heatmap_data.columns]
# Reorder columns by dataset
clr, dii = "CLR", "DII"
col_order_reordered = []
for ds in [clr, dii]:
col_order_reordered.extend([c for c in heatmap_data.columns if c.startswith(ds)])
heatmap_data = heatmap_data.loc[:, col_order_reordered]
# Find dataset boundaries for separator lines
boundaries = []
prev_dataset = None
for i, col in enumerate(col_order_reordered):
current_dataset = col.split("-motif")[0]
if prev_dataset is not None and current_dataset != prev_dataset:
boundaries.append(i)
prev_dataset = current_dataset
# Plot
fig, ax = plt.subplots(figsize=(max(8, len(col_order_reordered) * 0.8), max(4, len(heatmap_data) * 0.45)))
sns.heatmap(
heatmap_data, cmap="GnBu", linewidths=0.1, linecolor="lightgrey",
annot=True, fmt=".2f", annot_kws={"fontsize": 9},
cbar_kws={"label": "Frequency"}, ax=ax,
)
for b in boundaries:
ax.axvline(x=b, color="black", linewidth=1)
plt.title(f"Enriched motifs surrounding {anchor_ct} per subtype", fontsize=14, pad=20)
plt.ylabel("Cell type")
plt.xlabel("Motifs", fontsize=12)
plt.xticks(rotation=45, ha="right", fontsize=9)
plt.yticks(rotation=0, fontsize=10)
plt.tight_layout()
plt.show()
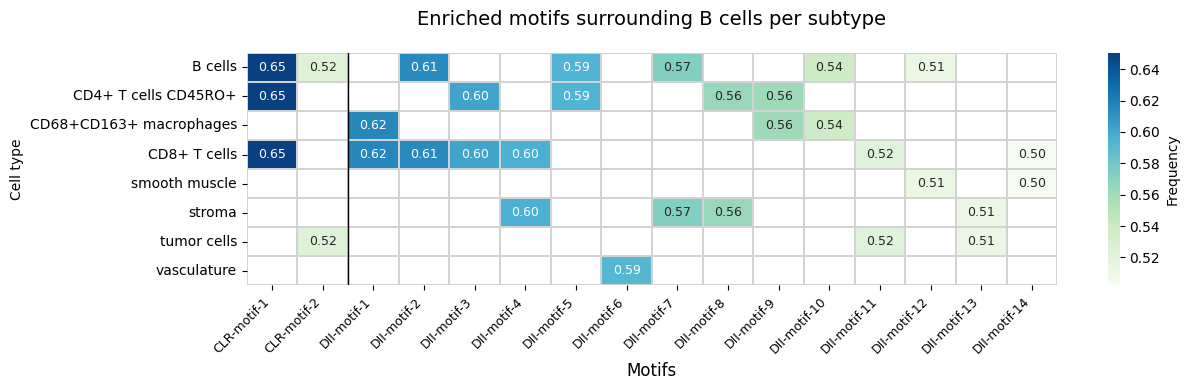
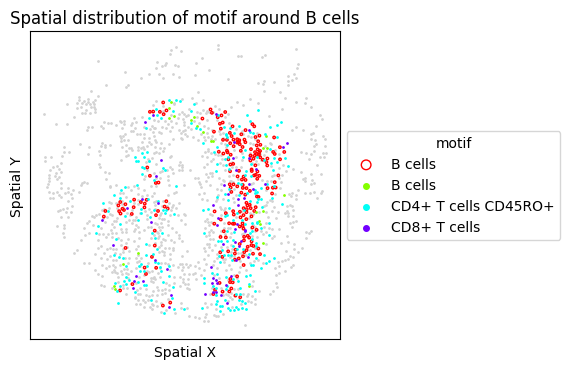
6. Targeted Motif Analysis: Tertiary Lymphoid Structures (TLS)
TLS are organized immune aggregates associated with anti-tumor immunity. We test a TLS-associated motif (B cells + T cell subsets) and visualize the FOVs with the highest motif frequency.
[13]:
motif_tls = ["B cells", "CD4+ T cells", "CD4+ T cells CD45RO+", "CD8+ T cells"]
motif_bcell, id_motif, id_center = spm.motif_enrichment_dist(
ct=anchor_ct,
motifs=motif_tls,
dataset=clr,
max_dist=max_dist,
return_cellID=True
)
[14]:
freq = {
clr: [],
}
freq_ds_names = {
clr: [],
}
for sp in spm.spatial_queries:
sp_ds_name = sp.dataset.split('_')[0]
if sp_ds_name == clr:
if anchor_ct not in sp.labels.values:
continue
out = sp.motif_enrichment_dist(
ct=anchor_ct,
motifs=motif_tls,
max_dist=max_dist,
return_cellID=False
)
if len(out) == 0:
continue
ratio = out['n_center_motif'] / out['n_center']
if isinstance(ratio, (pd.Series, np.ndarray)):
ratio = ratio.iloc[0] if hasattr(ratio, "iloc") else ratio.item()
ratio = float(ratio)
freq[sp_ds_name].append(ratio)
freq_ds_names[sp_ds_name].append(sp.dataset)
Found no ['CD4+ T cells'] in ClusterName. Ignoring them.
Found no ['CD4+ T cells'] in ClusterName. Ignoring them.
Found no ['CD4+ T cells'] in ClusterName. Ignoring them.
Found no ['CD4+ T cells'] in ClusterName. Ignoring them.
Found no ['CD4+ T cells'] in ClusterName. Ignoring them.
Found no ['CD4+ T cells'] in ClusterName. Ignoring them.
Found no ['CD4+ T cells'] in ClusterName. Ignoring them.
Found no ['CD4+ T cells'] in ClusterName. Ignoring them.
Found no ['CD4+ T cells'] in ClusterName. Ignoring them.
Found no ['CD4+ T cells'] in ClusterName. Ignoring them.
... (16 lines omitted) ...
Found no ['CD4+ T cells'] in ClusterName. Ignoring them.
Found no ['CD4+ T cells'] in ClusterName. Ignoring them.
[15]:
def get_top3_names(freq, freq_ds_names, k=3):
"""Return dataset names of the top-k FOVs by motif frequency."""
result = {}
for key in freq:
paired = sorted(zip(freq[key], freq_ds_names[key]), reverse=True)[:k]
result[key] = [name for _, name in paired]
return result
top3 = get_top3_names(freq, freq_ds_names)
top3
[15]:
{'CLR': ['CLR_20', 'CLR_14', 'CLR_28']}
[16]:
for sp in spm.spatial_queries:
if sp.dataset in top3[clr]:
sp.plot_motif_celltype(
ct=anchor_ct,
motif=motif_tls,
max_dist=max_dist,
figsize=(4, 4),
)
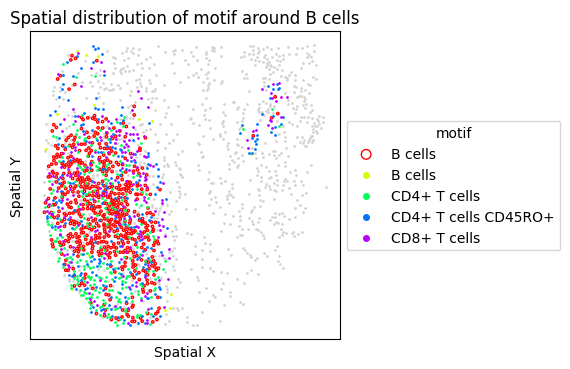
Found no ['CD4+ T cells'] in ClusterName. Ignoring them.
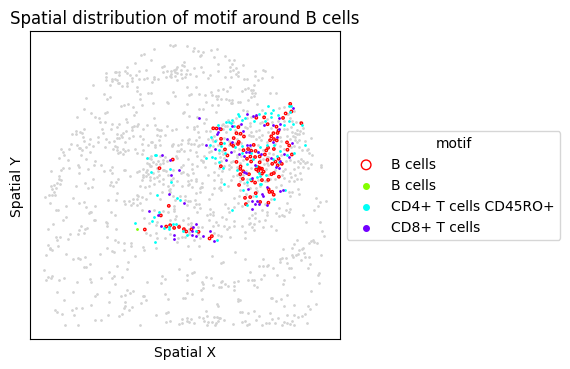
Found no ['CD4+ T cells'] in ClusterName. Ignoring them.
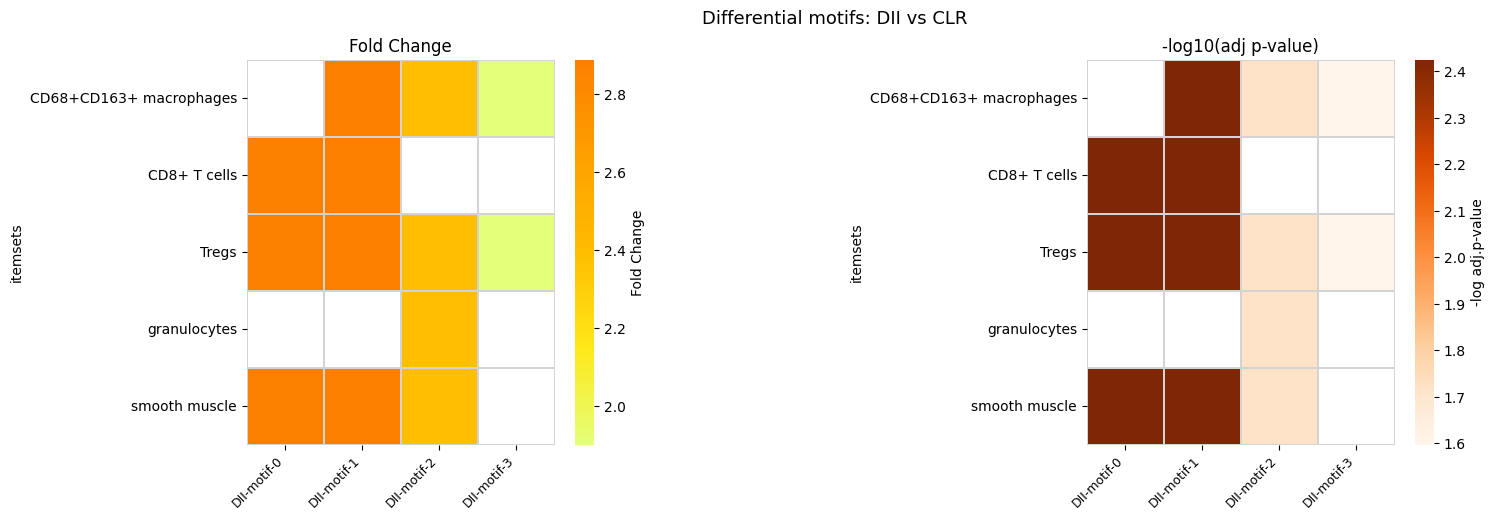
7. Differential Motif Analysis
Identify motifs whose frequency differs significantly between CLR and DII subtypes using differential_analysis_dist.
[17]:
diff_motif_bcell = spm.differential_analysis_dist(
ct=anchor_ct,
datasets=[clr, dii],
max_dist=max_dist,
min_support=min_support
)
Discovered 501 motifs across the datasets for differential analysis.
[18]:
diff_motif_bcell[clr]
[18]:
| itemsets | support_CLR_mean | support_DII_mean | adj-pval |
|---|
[19]:
diff_motif_bcell[dii]
[19]:
| itemsets | support_CLR_mean | support_DII_mean | adj-pval | |
|---|---|---|---|---|
| 0 | (CD8+ T cells, Tregs, smooth muscle) | 0.050989 | 0.146879 | 0.003753 |
| 1 | (CD68+CD163+ macrophages, CD8+ T cells, Tregs,... | 0.048059 | 0.138878 | 0.003753 |
| 2 | (CD68+CD163+ macrophages, Tregs, granulocytes,... | 0.041341 | 0.098820 | 0.019282 |
| 3 | (CD68+CD163+ macrophages, Tregs) | 0.159009 | 0.302162 | 0.025271 |
[20]:
diff_motif_bcell[clr]['group'] = clr
diff_motif_bcell[dii]['group'] = dii
enrich = diff_motif_bcell[dii]
enrich = pd.concat([enrich, diff_motif_bcell[clr]], axis=0)
enrich['fold_change'] = enrich[f'support_{dii}_mean'] / enrich[f'support_{clr}_mean']
enrich['motif_group'] = enrich['group'] + '_motif_' + enrich.index.astype(str)
col_order = enrich['motif_group'].tolist()
enrich_expanded = enrich.explode('itemsets')
heatmap_fc = enrich_expanded.pivot_table(
index='itemsets', columns='motif_group', values='fold_change'
)[col_order]
heatmap_pval = enrich_expanded.pivot_table(
index='itemsets', columns='motif_group', values='adj-pval'
)
heatmap_pval = -np.log10(heatmap_pval)[col_order]
col_labels = [c.replace('_', '-') for c in col_order]
heatmap_fc.columns = col_labels
heatmap_pval.columns = col_labels
fig, (ax1, ax2) = plt.subplots(1, 2, figsize=(16, 5), gridspec_kw={'wspace': 1.1})
sns.heatmap(heatmap_fc, cmap='Wistia', linewidths=0.1, linecolor='lightgrey',
square=True, cbar_kws={'label': 'Fold Change'}, ax=ax1)
ax1.set_title('Fold Change')
ax1.set_xlabel('')
ax1.set_xticklabels(ax1.get_xticklabels(), rotation=45, ha='right', fontsize=9)
sns.heatmap(heatmap_pval, cmap='Oranges', linewidths=0.1, linecolor='lightgrey',
square=True, cbar_kws={'label': '-log adj.p-value'}, ax=ax2)
ax2.set_title('-log10(adj p-value)')
ax2.set_xlabel('')
ax2.set_xticklabels(ax2.get_xticklabels(), rotation=45, ha='right', fontsize=9)
fig.suptitle(f'Differential motifs: {dii} vs {clr}', fontsize=13)
plt.tight_layout()
plt.show()
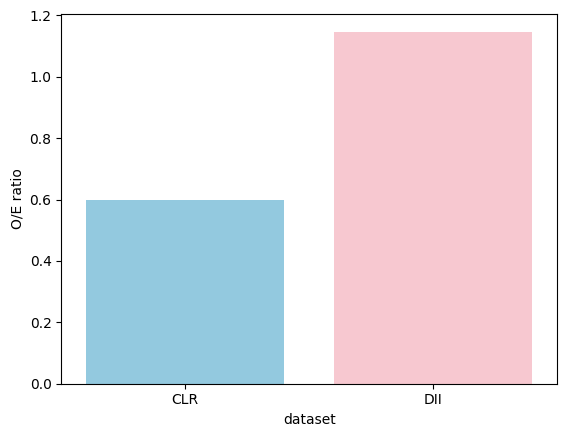
8. Targeted Motif Analysis: Immunosuppressive Niche
Test a hypothesis-driven immunosuppressive motif (macrophages + Tregs + CD8+ T cells + granulocytes) and compare its enrichment between CLR and DII.
[21]:
motif_immsup = ["CD68+CD163+ macrophages", "Tregs", "CD8+ T cells", "granulocytes"]
immsup_clr = spm.motif_enrichment_dist(ct=anchor_ct, motifs=motif_immsup, max_dist=max_dist, dataset=clr)
immsup_dii = spm.motif_enrichment_dist(ct=anchor_ct, motifs=motif_immsup, max_dist=max_dist, dataset=dii)
immsup_clr["dataset"] = clr
immsup_dii["dataset"] = dii
immsup_both = pd.concat([immsup_clr, immsup_dii], axis=0)
immsup_both["O/E ratio"] = immsup_both["n_center_motif"] / immsup_both["expectation"]
immsup_both
[21]:
| center | motifs | n_center_motif | n_center | n_motif | expectation | p-values | if_significant | dataset | O/E ratio | |
|---|---|---|---|---|---|---|---|---|---|---|
| 0 | B cells | [CD68+CD163+ macrophages, CD8+ T cells, Tregs,... | 384 | 10269 | 6949 | 642.523307 | 1.000000 | False | CLR | 0.597644 |
| 0 | B cells | [CD68+CD163+ macrophages, CD8+ T cells, Tregs,... | 380 | 2774 | 17593 | 331.262944 | 0.002119 | True | DII | 1.147125 |
[22]:
sns.barplot(x="dataset", y="O/E ratio", data=immsup_both, palette=["skyblue", "pink"])
plt.show()
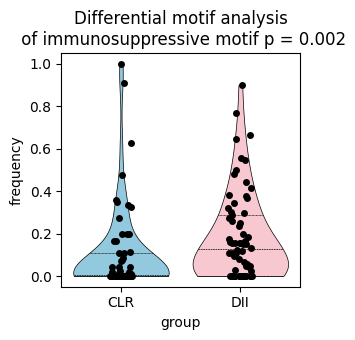
Compute per-FOV motif frequency to test whether the immunosuppressive motif is differentially present between CLR and DII (Mann-Whitney U test).
[23]:
freq = {clr: [], dii: []}
for sp in spm.spatial_queries:
sp_ds_name = sp.dataset.split("_")[0]
if anchor_ct not in sp.labels.values:
continue
out = sp.motif_enrichment_dist(
ct=anchor_ct, motifs=motif_immsup, max_dist=max_dist, return_cellID=False,
)
if len(out) == 0:
continue
ratio = out["n_center_motif"] / out["n_center"]
if isinstance(ratio, (pd.Series, np.ndarray)):
ratio = ratio.iloc[0] if hasattr(ratio, "iloc") else ratio.item()
freq[sp_ds_name].append(float(ratio))
Found no ['Tregs'] in ClusterName. Ignoring them.
Found no ['Tregs'] in ClusterName. Ignoring them.
Found no ['Tregs'] in ClusterName. Ignoring them.
Found no ['Tregs'] in ClusterName. Ignoring them.
Found no ['Tregs'] in ClusterName. Ignoring them.
Found no ['Tregs'] in ClusterName. Ignoring them.
Found no ['Tregs'] in ClusterName. Ignoring them.
[24]:
plot_df = pd.DataFrame({
'group': (
[clr] * len(freq[clr]) +
[dii] * len(freq[dii])
),
'frequency': (
freq[clr] +
freq[dii]
)
})
from scipy import stats
group1 = freq[clr]
group2 = freq[dii]
stat, p_value = stats.mannwhitneyu(group1, group2,
alternative='two-sided',
method='auto')
fig, ax = plt.subplots(figsize=(3.2, 3.5))
# Violin
sns.violinplot(
data=plot_df,
x="group",
y="frequency",
ax=ax,
cut=0,
inner="quartile",
linewidth=0.5,
edgecolor='black',
palette=["skyblue", "pink"]
)
# Overlay points
sns.stripplot(
data=plot_df,
x="group",
y="frequency",
ax=ax,
color="black",
size=5,
jitter=0.1,
)
ax.set_title(f'Differential motif analysis\n of immunosuppressive motif p = {p_value:.3f}')
plt.tight_layout()
[ ]: