Multi-dataset: Gene-Gene Covariation Analysis
This example demonstrates how to identify cross-cell gene-gene covariation across multiple FOVs.
The multi-FOV version pools anchor–neighbor cell pairs across all FOVs within a condition, increasing statistical power for detecting spatially-specific gene correlations.
Dataset: CZI Kidney — normal vs autosomal dominant tubulointerstitial kidney disease (ADTKD) (spatial transcriptomics), availabel at https://cellxgene.cziscience.com/collections/8e880741-bf9a-4c8e-9227-934204631d2a
Key API:
spatial_query_multi.compute_gene_gene_correlation— pools all non-anchor motif cell types togetherspatial_query_multi.compute_gene_gene_correlation_by_type— tests each non-anchor cell type separately
Tip: For whole-transcriptomic data like this kidney dataset, we’d suggest pre-selecting highly variable genes via the
genesparameter to improve speed and reduce multiple-testing burden.
Setup
[1]:
import warnings
warnings.filterwarnings("ignore")
import os
import anndata as ad
import scanpy as sc
import numpy as np
import matplotlib.pyplot as plt
from SpatialQuery import spatial_query_multi
Load Data & Initialize
[3]:
DATA_DIR = "../data/CZI_kidney"
data_files = os.listdir(DATA_DIR)
adatas = [ad.read_h5ad(os.path.join(DATA_DIR, f)) for f in data_files]
print(f"Loaded {len(adatas)} FOVs")
adatas[0]
Loaded 84 FOVs
[3]:
AnnData object with n_obs × n_vars = 12906 × 17811
obs: 'assay_ontology_term_id', 'self_reported_ethnicity_ontology_term_id', 'is_primary_data', 'organism_ontology_term_id', 'sample', 'tissue_ontology_term_id', 'disease_state', 'sex_ontology_term_id', 'genotype', 'development_stage_ontology_term_id', 'author_cell_type', 'cell_type_ontology_term_id', 'disease_ontology_term_id', 'donor_id', 'suspension_type', 'tissue_type', 'cell_type', 'assay', 'disease', 'organism', 'sex', 'tissue', 'self_reported_ethnicity', 'development_stage', 'observation_joinid'
var: 'feature_is_filtered', 'feature_name', 'feature_reference', 'feature_biotype', 'feature_length'
uns: 'citation', 'schema_reference', 'schema_version', 'title'
obsm: 'X_spatial'
[4]:
# Create condition labels and filter out mitochondrial / unannotated genes
for i, adata in enumerate(adatas):
adata.obs['disease_state_genotype'] = (
adata.obs['disease_state'].astype(str) + '_' + adata.obs['genotype'].astype(str)
)
adata.var_names = adata.var['feature_name'].tolist()
adata = adata[:, ~adata.var_names.str.startswith('mt-')]
adata = adata[:, ~adata.var_names.str.endswith('Rik')]
adata = adata[:, ~adata.var_names.str.startswith('Gm')]
adatas[i] = adata
[5]:
# Map verbose condition names to short labels
dataset_mapping = {
'early diabetic kidney disease_BTBR-ob/ob': 'DKD_BTBR-ob/ob',
'autosomal dominant tubulointersital kidney disease (ADTKD)_UMOD-KI/KI': 'ADTKD_UMOD-KI/KI',
'control_BTBR-wt/wt': 'control_BTBR-WT/WT',
'control_UMOD-WT/WT': 'control_UMOD-WT/WT',
}
dataset_name_col = 'disease_state_genotype'
datasets = [adata.obs[dataset_name_col].unique()[0] for adata in adatas]
dataset_names = [dataset_mapping[d] for d in datasets]
[6]:
spatial_key = "X_spatial"
label_key = "cell_type"
feature_name = 'feature_name'
[7]:
spm = spatial_query_multi(
adatas=adatas,
datasets=dataset_names,
spatial_key=spatial_key,
label_key=label_key,
feature_name=feature_name,
build_gene_index=False,
if_lognorm=True,
if_normalize_spatial_coord=True
)
Replacing _ with hyphen in DKD_BTBR-ob/ob.
Replacing _ with hyphen in DKD_BTBR-ob/ob.
Replacing _ with hyphen in control_BTBR-WT/WT.
Replacing _ with hyphen in control_UMOD-WT/WT.
Replacing _ with hyphen in ADTKD_UMOD-KI/KI.
Replacing _ with hyphen in ADTKD_UMOD-KI/KI.
Replacing _ with hyphen in ADTKD_UMOD-KI/KI.
Replacing _ with hyphen in control_BTBR-WT/WT.
Replacing _ with hyphen in control_BTBR-WT/WT.
Replacing _ with hyphen in DKD_BTBR-ob/ob.
...(492 lines omitted)...
build_gene_index is False. Using adata.X for gene expression analysis.
Log normalizing the expression data... If data is already log normalized, please set if_lognorm to False.
Select Highly Variable Genes
For whole-transcriptomic data, pre-selecting highly variable genes (HVGs) speeds up computation and focuses the analysis on the most informative features.
[8]:
valid_ds_names = list(set(s.dataset.split('_')[0] for s in spm.spatial_queries))
valid_ds_names
[8]:
['DKD-BTBR-ob/ob',
'ADTKD-UMOD-KI/KI',
'control-BTBR-WT/WT',
'control-UMOD-WT/WT']
[9]:
# Select top 3000 HVGs per condition and take the union
tt1 = np.array([f.replace('_','-') for f in dataset_names])
selected_genes = {}
tt2 = []
for adata in adatas:
adata.var_names = adata.var[feature_name]
tt2.append(adata)
for ds in valid_ds_names:
mask = np.where(tt1 == ds)[0]
adata_sub = ad.concat([tt2[i].copy() for i in mask], join='inner')
print(f"{ds}: {adata_sub.shape}")
sc.pp.normalize_total(adata_sub)
sc.pp.log1p(adata_sub)
sc.pp.highly_variable_genes(adata_sub, n_top_genes=3000)
selected_genes[ds] = adata_sub.var[adata_sub.var['highly_variable']].index.tolist()
DKD-BTBR-ob/ob: (613317, 13586)
ADTKD-UMOD-KI/KI: (501021, 14390)
control-BTBR-WT/WT: (550106, 13751)
control-UMOD-WT/WT: (336157, 14271)
Define Anchor and Motif
We use macrophage as the anchor cell type. The motif includes fibroblast and thick ascending limb epithelial cell.
[10]:
anchor_ct = "macrophage"
motif = ['kidney interstitial fibroblast', 'kidney loop of Henle thick ascending limb epithelial cell']
max_dist = 5
ds_control = 'control-UMOD-WT/WT'
ds_case = 'ADTKD-UMOD-KI/KI'
Method 1: Pooled Covariation (compute_gene_gene_correlation)
compute_gene_gene_correlation pools all non-anchor cell types in the motif into a single neighbor group before computing correlations across FOVs. It performs two tests:
Test 1 (spatial specificity): correlation in motif+ anchor–neighbor pairs vs randomly paired cells
Test 2 (motif specificity): correlation in motif+ pairs vs motif− anchor–neighbor pairs
Key parameters:
Parameter |
Description |
|---|---|
|
Anchor cell type |
|
List of neighbor cell types forming the motif |
|
Condition name(s) to restrict the analysis to |
|
Neighborhood radius (use either |
|
Number of nearest neighbors (use either |
|
List of genes to analyze. If |
|
Minimum non-zero expression values required to include a gene |
|
FDR significance threshold (default: 0.05) |
Key output columns:
Column |
Description |
|---|---|
|
Gene expressed in the anchor cell |
|
Gene expressed in the neighbor cell |
|
Correlation between motif+ anchor–neighbor pairs |
|
Correlation between randomly paired cells (Test 1 baseline) |
|
Correlation for motif− anchors (Test 2 baseline) |
|
Combined effect size from both tests |
|
Whether the pair passes both FDR-corrected tests |
[11]:
covarying_pooled = spm.compute_gene_gene_correlation(
ct=anchor_ct,
motif=motif,
dataset=ds_case,
max_dist=max_dist,
genes=selected_genes[ds_case],
alpha=0.05,
)
covarying_pooled_sig = covarying_pooled[covarying_pooled["if_significant"]].copy()
print(f"Total gene pairs tested: {len(covarying_pooled)}")
print(f"Significant covarying pairs (pooled): {len(covarying_pooled_sig)}")
covarying_pooled_sig.head(10)
Computing covarying genes using expression data ...
Gene coverage: 14390 genes in all FOVs, 18795 genes total (union)
-> 4405 genes present in subset of FOVs (will use available data)
Analyzing 3000 genes across 24 FOVs
================================================================================
Step 1: Computing and accumulating statistics across FOVs
================================================================================
--- Processing FOV 1/24: ADTKD-UMOD-KI/KI_0 ---
...(177 lines omitted)...
Total gene pairs tested: 9000000
Significant covarying pairs (pooled): 296
[11]:
| gene_center | gene_motif | corr_neighbor | corr_non_neighbor | p_value_test1 | delta_corr_test1 | corr_center_no_motif | p_value_test2 | delta_corr_test2 | combined_score | adj-pval-test1 | adj-pval-test2 | if_significant | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | Wif1 | Wif1 | 0.312311 | -0.011133 | 0.0 | 0.323444 | -0.000243 | 0.0 | 0.312554 | 94.746418 | 0.0 | 0.0 | True |
| 1 | Armc12 | Wif1 | 0.300108 | -0.010945 | 0.0 | 0.311053 | -0.000126 | 0.0 | 0.300234 | 91.043878 | 0.0 | 0.0 | True |
| 2 | Grid1 | Grin3a | 0.259830 | 0.000530 | 0.0 | 0.259300 | -0.000535 | 0.0 | 0.260365 | 78.013763 | 0.0 | 0.0 | True |
| 3 | Hpdl | Dnah3 | 0.236931 | -0.001030 | 0.0 | 0.237962 | 0.000073 | 0.0 | 0.236859 | 71.156931 | 0.0 | 0.0 | True |
| 4 | Grid1 | Myom2 | 0.221014 | -0.000439 | 0.0 | 0.221453 | 0.000255 | 0.0 | 0.220759 | 66.290110 | 0.0 | 0.0 | True |
| 5 | Cercam | Rergl | 0.212219 | -0.000659 | 0.0 | 0.212879 | 0.004272 | 0.0 | 0.207947 | 62.828013 | 0.0 | 0.0 | True |
| 6 | Kap | Kap | 0.368871 | -0.012893 | 0.0 | 0.381763 | 0.251811 | 0.0 | 0.117059 | 58.941187 | 0.0 | 0.0 | True |
| 7 | Cyp2d12 | Hmx2 | 0.192851 | -0.000510 | 0.0 | 0.193361 | 0.001287 | 0.0 | 0.191563 | 57.630835 | 0.0 | 0.0 | True |
| 8 | Ptprr | Tmc7 | 0.191837 | -0.000012 | 0.0 | 0.191850 | 0.003037 | 0.0 | 0.188800 | 56.914508 | 0.0 | 0.0 | True |
| 9 | Lypd1 | Notumos | 0.180410 | -0.000194 | 0.0 | 0.180604 | -0.000311 | 0.0 | 0.180721 | 54.205766 | 0.0 | 0.0 | True |
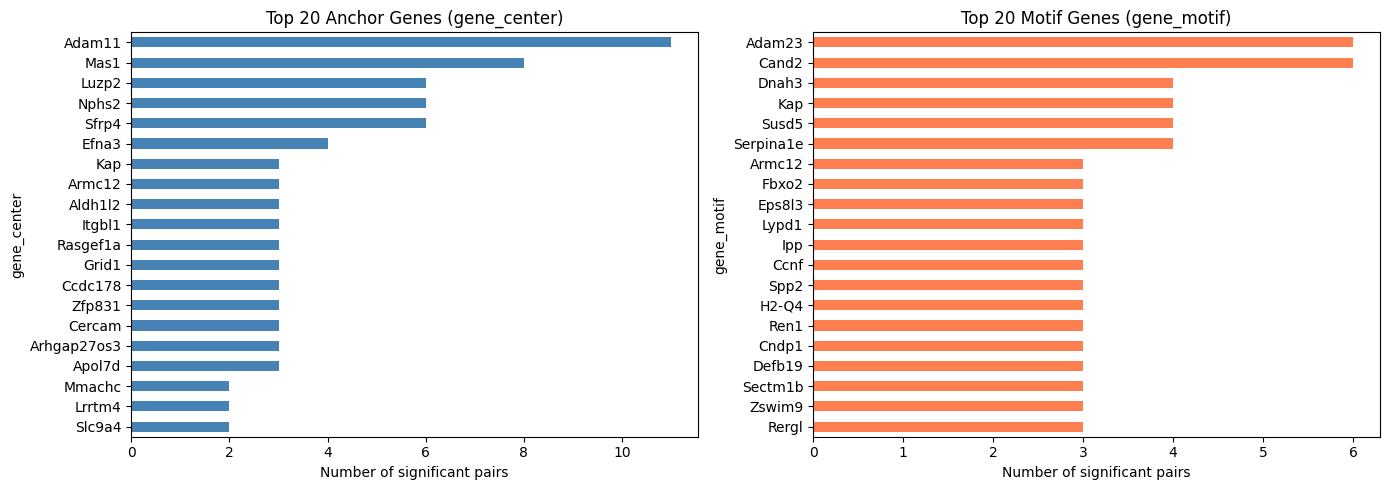
Top Frequent Genes in Covarying Pairs (Pooled)
Visualize the top 20 most frequently appearing genes on the anchor side (gene_center) and the motif side (gene_motif) among significant covarying pairs.
[12]:
top_n = 20
fig, axes = plt.subplots(1, 2, figsize=(14, 5))
covarying_pooled_sig["gene_center"].value_counts().head(top_n).plot.barh(
ax=axes[0], color="steelblue")
axes[0].set_title(f"Top {top_n} Anchor Genes (gene_center)")
axes[0].set_xlabel("Number of significant pairs")
axes[0].invert_yaxis()
covarying_pooled_sig["gene_motif"].value_counts().head(top_n).plot.barh(
ax=axes[1], color="coral")
axes[1].set_title(f"Top {top_n} Motif Genes (gene_motif)")
axes[1].set_xlabel("Number of significant pairs")
axes[1].invert_yaxis()
plt.tight_layout()
plt.show()
Method 2: Per-cell-type Covariation (compute_gene_gene_correlation_by_type)
compute_gene_gene_correlation_by_type tests each non-anchor cell type separately, so the output includes a cell_type column.
Key parameters:
Parameter |
Description |
|---|---|
|
Anchor cell type |
|
List of neighbor cell types forming the motif |
|
Condition name(s) to restrict the analysis to. If |
|
List of genes to analyze. If |
|
Neighborhood radius (use either |
|
Number of nearest neighbors (use either |
|
Minimum neighborhood size for each anchor cell |
|
Minimum non-zero expression values required to include a gene |
|
FDR significance threshold (default: 0.05) |
Key output columns:
Column |
Description |
|---|---|
|
The neighbor cell type in the pair |
|
Gene expressed in the anchor cell |
|
Gene expressed in the neighbor cell |
|
Correlation between motif+ anchor–neighbor pairs |
|
Correlation between randomly paired cells (Test 1 baseline) |
|
Correlation for motif− anchors (Test 2 baseline) |
|
Combined effect size from both tests |
|
Whether the pair passes both FDR-corrected tests |
[13]:
covarying = spm.compute_gene_gene_correlation_by_type(
ct=anchor_ct,
motif=motif,
dataset=ds_case,
max_dist=max_dist,
genes=selected_genes[ds_case],
alpha=0.05,
)
covarying_sig = covarying[covarying["if_significant"]].copy()
print(f"Total gene pairs tested: {len(covarying)}")
print(f"Significant covarying pairs: {len(covarying_sig)}")
Computing covarying genes using expression data ...
Analyzing 2 non-center cell types in motif: ['kidney interstitial fibroblast', 'kidney loop of Henle thick ascending limb epithelial cell']
================================================================================
Selected 24 FOVs for analysis
Gene coverage: 14390 genes in all FOVs, 18795 genes total (union)
-> 4405 genes present in subset of FOVs (will use available data)
Analyzing 3000 genes across 24 FOVs
================================================================================
Step 1: Computing Correlation-3 (Center without motif vs Neighbors)
...(137 lines omitted)...
Total gene pairs tested: 15536862
Significant covarying pairs: 3501
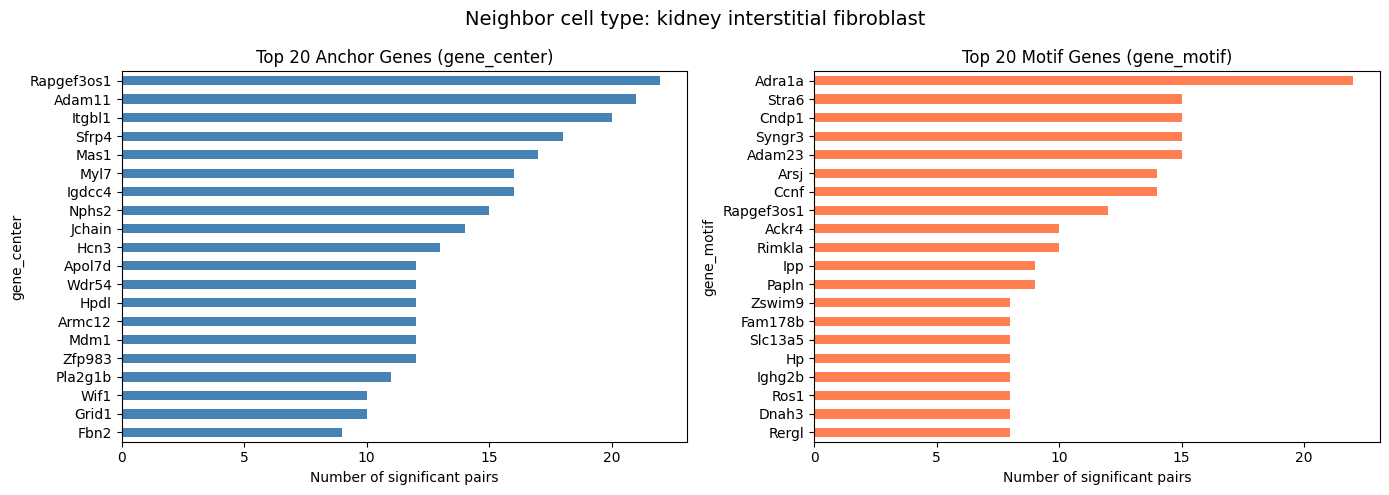
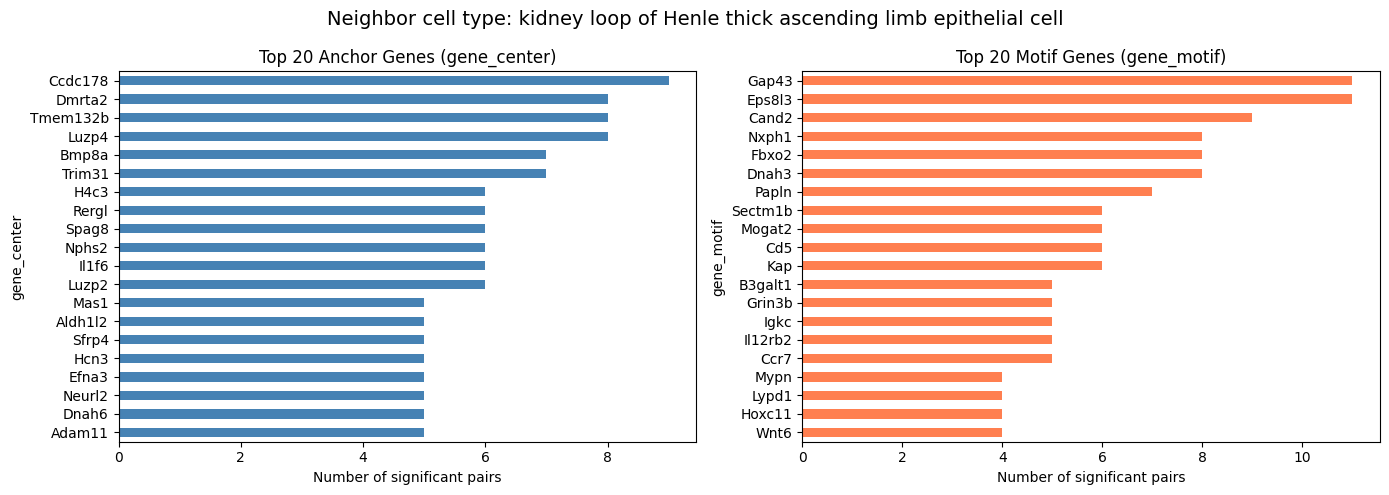
Top Frequent Genes in Covarying Pairs (Per Cell Type)
For compute_gene_gene_correlation_by_type, visualize the top 20 genes separately for each neighbor cell type.
[14]:
top_n = 20
cell_types = covarying_sig["cell_type"].unique()
for ct in cell_types:
ct_df = covarying_sig[covarying_sig["cell_type"] == ct]
if len(ct_df) == 0:
continue
fig, axes = plt.subplots(1, 2, figsize=(14, 5))
fig.suptitle(f"Neighbor cell type: {ct}", fontsize=14)
ct_df["gene_center"].value_counts().head(top_n).plot.barh(
ax=axes[0], color="steelblue")
axes[0].set_title(f"Top {top_n} Anchor Genes (gene_center)")
axes[0].set_xlabel("Number of significant pairs")
axes[0].invert_yaxis()
ct_df["gene_motif"].value_counts().head(top_n).plot.barh(
ax=axes[1], color="coral")
axes[1].set_title(f"Top {top_n} Motif Genes (gene_motif)")
axes[1].set_xlabel("Number of significant pairs")
axes[1].invert_yaxis()
plt.tight_layout()
plt.show()
Summary by Neighbor Cell Type
[15]:
covarying_sig["cell_type"].value_counts()
[15]:
cell_type
kidney interstitial fibroblast 2525
kidney loop of Henle thick ascending limb epithelial cell 976
Name: count, dtype: int64
Inspect Top Covarying Gene Pairs
Sort by combined score (absolute value) to see the strongest associations.
[16]:
covarying_sig.sort_values("abs_combined_score", ascending=False).head(10)
[16]:
| cell_type | gene_center | gene_motif | corr_neighbor | corr_non_neighbor | p_value_test1 | delta_corr_test1 | corr_center_no_motif | p_value_test2 | delta_corr_test2 | combined_score | q_value_test1 | q_value_test2 | reject_test1_fdr | reject_test2_fdr | abs_combined_score | if_significant | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | kidney interstitial fibroblast | Wif1 | Lmo3 | 0.815627 | -0.020971 | 0.0 | 0.836598 | -0.000371 | 0.0 | 0.815997 | 246.653183 | 0.0 | 0.0 | True | True | 246.653183 | True |
| 1 | kidney interstitial fibroblast | Wif1 | Wif1 | 0.786223 | -0.035668 | 0.0 | 0.821892 | -0.000243 | 0.0 | 0.786467 | 239.128233 | 0.0 | 0.0 | True | True | 239.128233 | True |
| 2 | kidney interstitial fibroblast | Armc12 | Lmo3 | 0.748789 | -0.020264 | 0.0 | 0.769053 | -0.000078 | 0.0 | 0.748867 | 226.476755 | 0.0 | 0.0 | True | True | 226.476755 | True |
| 3 | kidney interstitial fibroblast | Rapgef3os1 | Slamf1 | 0.742925 | -0.017139 | 0.0 | 0.760064 | -0.000098 | 0.0 | 0.743023 | 224.440595 | 0.0 | 0.0 | True | True | 224.440595 | True |
| 4 | kidney interstitial fibroblast | Armc12 | Wif1 | 0.721795 | -0.034466 | 0.0 | 0.756261 | -0.000126 | 0.0 | 0.721921 | 219.666989 | 0.0 | 0.0 | True | True | 219.666989 | True |
| 5 | kidney interstitial fibroblast | Luzp2 | Serpina1e | 0.699316 | -0.024935 | 0.0 | 0.724251 | 0.005572 | 0.0 | 0.693744 | 210.868927 | 0.0 | 0.0 | True | True | 210.868927 | True |
| 6 | kidney interstitial fibroblast | Rapgef3os1 | Cerox1 | 0.671484 | -0.019855 | 0.0 | 0.691338 | -0.000087 | 0.0 | 0.671571 | 203.250420 | 0.0 | 0.0 | True | True | 203.250420 | True |
| 7 | kidney interstitial fibroblast | Fam178b | Ppp1r3d | 0.601521 | -0.000445 | 0.0 | 0.601967 | -0.000137 | 0.0 | 0.601659 | 180.525313 | 0.0 | 0.0 | True | True | 180.525313 | True |
| 8 | kidney interstitial fibroblast | Gap43 | Best3 | 0.528982 | -0.016956 | 0.0 | 0.545937 | -0.000250 | 0.0 | 0.529231 | 160.272942 | 0.0 | 0.0 | True | True | 160.272942 | True |
| 9 | kidney interstitial fibroblast | Cercam | Rergl | 0.493953 | -0.001935 | 0.0 | 0.495888 | 0.004272 | 0.0 | 0.489681 | 147.462958 | 0.0 | 0.0 | True | True | 147.462958 | True |
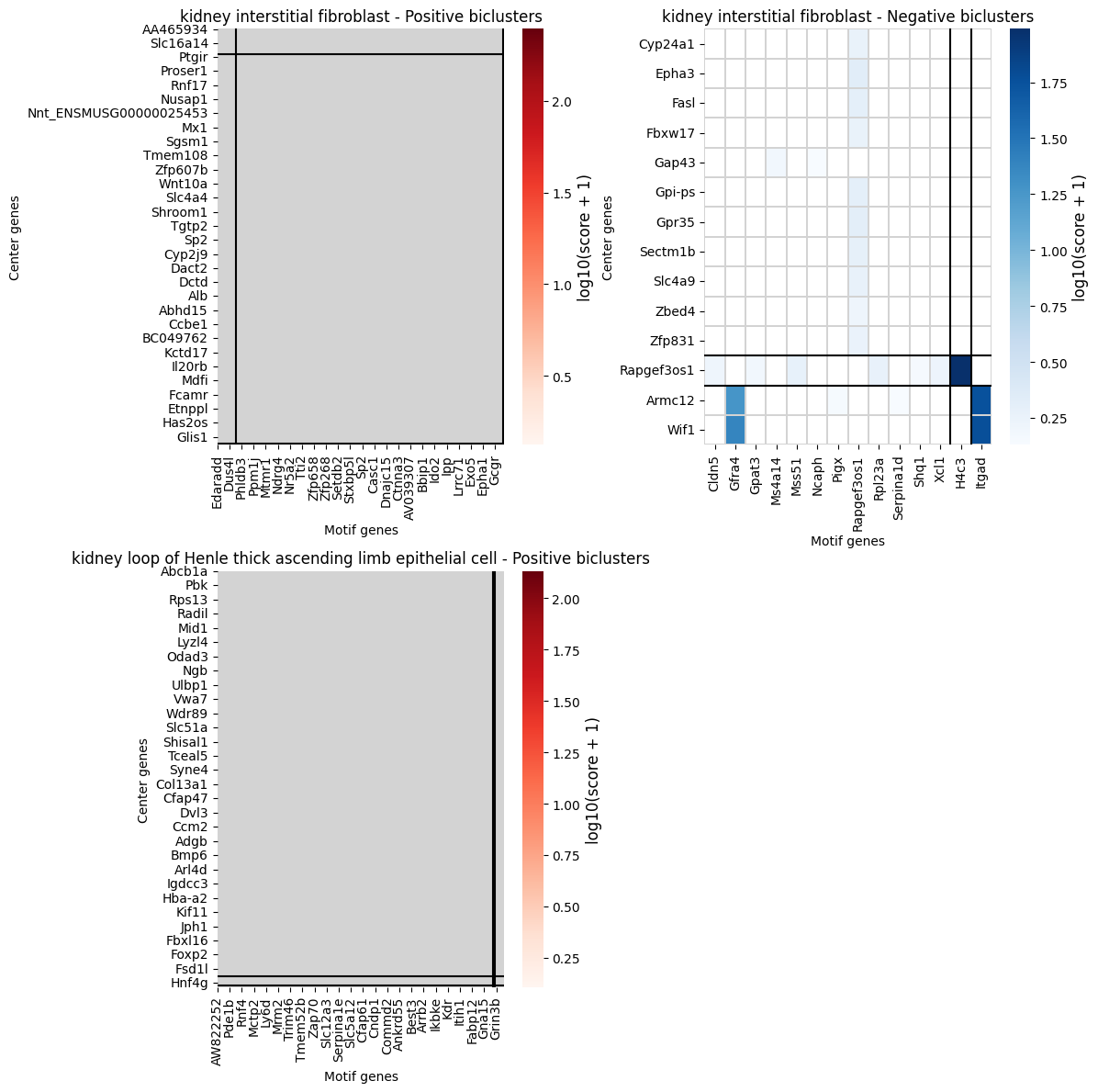
Visualize Covarying Gene Modules
Note:
plot_gene_pair_heatmapis available on the single-datasetspatial_queryclass. For multi-dataset results, you can use the standalone plotting function fromSpatialQuery.plottingor apply clustering directly to the result DataFrame.
[17]:
from SpatialQuery.plotting import plot_gene_pair_heatmap
if len(covarying_sig) > 0:
modules = plot_gene_pair_heatmap(
gene_pair_df=covarying_sig,
figsize=(12, 6),
)
else:
print("No significant covarying gene pairs found.")
kidney loop of Henle thick ascending limb epithelial cell: Skipping neg with 4 rows and 1 columns.
[18]:
modules
[18]:
| gene_center | gene_motif | combined_score | cluster_type | cluster_row | cluster_col | cell_type | |
|---|---|---|---|---|---|---|---|
| 0 | AA465934 | Adamts7 | 25.321869 | positive | 0 | 1 | kidney interstitial fibroblast |
| 1 | AA465934 | Il12rb2 | 26.951418 | positive | 0 | 1 | kidney interstitial fibroblast |
| 2 | AA465934 | Tmem143 | 1.009324 | positive | 0 | 1 | kidney interstitial fibroblast |
| 3 | AA914427 | Dgki | 0.660621 | positive | 1 | 1 | kidney interstitial fibroblast |
| 4 | AA914427 | Iqcm | 0.763277 | positive | 1 | 1 | kidney interstitial fibroblast |
| ... | ... | ... | ... | ... | ... | ... | ... |
| 3492 | Zfp983 | Eps8l3 | 42.379554 | positive | 0 | 2 | kidney loop of Henle thick ascending limb epit... |
| 3493 | Zfp991 | Nuggc | 0.338234 | positive | 0 | 0 | kidney loop of Henle thick ascending limb epit... |
| 3494 | Zfp992 | B3galt1 | 0.399425 | positive | 0 | 0 | kidney loop of Henle thick ascending limb epit... |
| 3495 | Zfp992 | Sectm1b | 0.568542 | positive | 0 | 0 | kidney loop of Henle thick ascending limb epit... |
| 3496 | Zkscan5 | Mypn | 0.492021 | positive | 0 | 0 | kidney loop of Henle thick ascending limb epit... |
3497 rows × 7 columns
[ ]: