Multi-dataset: Differential Motif Analysis
This example demonstrates how to identify motifs that are differentially enriched between two experimental conditions using differential_analysis_dist.
While motif enrichment (previous example) discovers motifs within each condition independently, differential motif analysis directly compares motif frequencies between conditions using a Mann-Whitney U test across FOVs, with FDR correction for multiple testing.
Dataset: CODEX spatial proteomics — CLR vs DII immune subtypes Key API: spatial_query_multi.differential_analysis_dist and spatial_query_multi.differential_analysis_knn
Setup
[1]:
import warnings
warnings.filterwarnings("ignore")
import anndata as ad
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import seaborn as sns
from SpatialQuery import spatial_query_multi
Load Data & Initialize
[3]:
DATA_DIR = "../data/codex_cancer"
adata = ad.read_h5ad(f"{DATA_DIR}/codex_data.h5ad")
adata
[3]:
AnnData object with n_obs × n_vars = 258385 × 56
obs: 'CellID', 'ClusterID', 'EventID', 'File Name', 'Region', 'TMA_AB', 'TMA_12', 'Index in File', 'groups', 'patients', 'spots', 'cell_id', 'size:size', 'HOECHST1_Cyc_1_ch_1', 'DRAQ5_Cyc_23_ch_4', 'Profile_Homogeneity:Fiter1', 'ClusterSize', 'ClusterName', 'neighborhood10', 'CD4+ICOS+', 'CD4+Ki67+', 'CD4+PD-1+', 'CD68+CD163+ICOS+', 'CD68+CD163+Ki67+', 'CD68+CD163+PD-1+', 'CD68+ICOS+', 'CD68+Ki67+', 'CD68+PD-1+', 'CD8+ICOS+', 'CD8+Ki67+', 'CD8+PD-1+', 'Treg-ICOS+', 'Treg-Ki67+', 'Treg-PD-1+', 'neighborhood number final', 'neighborhood name'
var: 'marker_name', 'full_name', 'cell_type_annotation', 'cycle', 'channel'
uns: 'data_source', 'groups_mapping', 'n_markers', 'technology'
obsm: 'X_spatial_global', 'X_spatial_tile'
[4]:
adata.X.max()
[4]:
np.float32(54776.695)
Inspect adata.obsm for spatial coordinates, adata.obs for cell type labels and FOV identifiers, and adata.var for feature names. Then set the corresponding column names below.
[5]:
# Assign immune subtype labels
adata.obs["state"] = "CLR"
adata.obs["state"][adata.obs["groups"] == 2] = "DII"
# Feature-wise z-score normalization for protein expression
adata.X = (adata.X - adata.X.mean(axis=0)) / adata.X.std(axis=0)
[6]:
spatial_key = "X_spatial_tile"
label_key = "ClusterName"
feature_name = "marker_name"
fov_key = "File Name"
[7]:
# Split data by FOV and immune subtype
clr_data = adata[adata.obs["state"] == "CLR"]
dii_data = adata[adata.obs["state"] == "DII"]
clr_datas = [clr_data[clr_data.obs[fov_key] == f] for f in clr_data.obs[fov_key].unique()]
dii_datas = [dii_data[dii_data.obs[fov_key] == f] for f in dii_data.obs[fov_key].unique()]
ds_names = ["CLR"] * len(clr_datas) + ["DII"] * len(dii_datas)
adata_fovs = clr_datas + dii_datas
print(f"CLR FOVs: {len(clr_datas)}, DII FOVs: {len(dii_datas)}")
CLR FOVs: 68, DII FOVs: 72
[8]:
spm = spatial_query_multi(
adatas=adata_fovs,
datasets=ds_names,
spatial_key=spatial_key,
label_key=label_key,
feature_name=feature_name,
build_gene_index=False,
if_lognorm=False, # data is already z-score normalized
if_normalize_spatial_coord=True,
)
Auto-normalizing spatial coordinates: mean nearest neighbor distance = 1.0
Scale factor: 0.0448
build_gene_index is False. Using adata.X for gene expression analysis.
Auto-normalizing spatial coordinates: mean nearest neighbor distance = 1.0
Scale factor: 0.0311
build_gene_index is False. Using adata.X for gene expression analysis.
Auto-normalizing spatial coordinates: mean nearest neighbor distance = 1.0
... (548 lines omitted) ...
Scale factor: 0.0421
build_gene_index is False. Using adata.X for gene expression analysis.
Run Differential Motif Analysis
differential_analysis_dist compares motif frequencies between two conditions across FOVs. It first discovers all motifs present in either condition, then tests whether the per-FOV support (frequency) of each motif differs significantly.
Key parameters:
Parameter |
Description |
|---|---|
|
Anchor cell type |
|
List of exactly two condition names to compare |
|
Neighborhood radius |
|
Minimum support threshold for motif mining |
Return value: A dictionary with one key per condition. Each value is a DataFrame of motifs that are significantly more frequent in that condition.
Output columns:
Column |
Description |
|---|---|
|
The motif (frozenset of cell types) |
|
Mean frequency across FOVs in condition 1 |
|
Mean frequency across FOVs in condition 2 |
|
FDR-adjusted p-value (Mann-Whitney U) |
[9]:
anchor_ct = "B cells"
max_dist = 5
min_support = 0.5
clr = "CLR"
dii = "DII"
diff_motif = spm.differential_analysis_dist(
ct=anchor_ct,
datasets=[clr, dii],
max_dist=max_dist,
min_support=min_support,
)
Discovered 501 motifs across the datasets for differential analysis.
Inspect Results
Check which motifs are enriched in each condition.
[10]:
print(f"Motifs enriched in {clr}: {len(diff_motif[clr])}")
diff_motif[clr]
Motifs enriched in CLR: 0
[10]:
| itemsets | support_CLR_mean | support_DII_mean | adj-pval |
|---|
[11]:
print(f"Motifs enriched in {dii}: {len(diff_motif[dii])}")
diff_motif[dii]
Motifs enriched in DII: 4
[11]:
| itemsets | support_CLR_mean | support_DII_mean | adj-pval | |
|---|---|---|---|---|
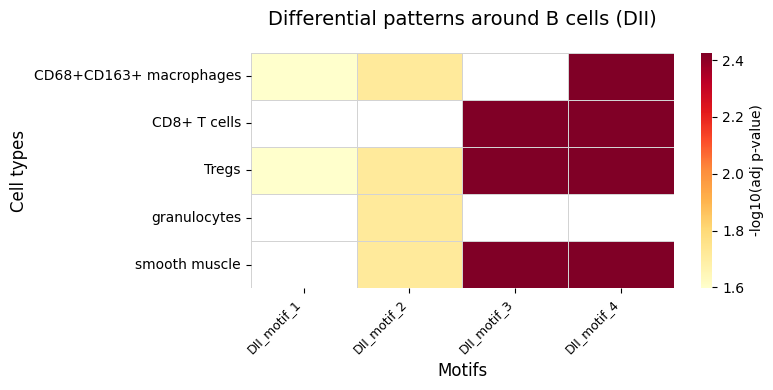
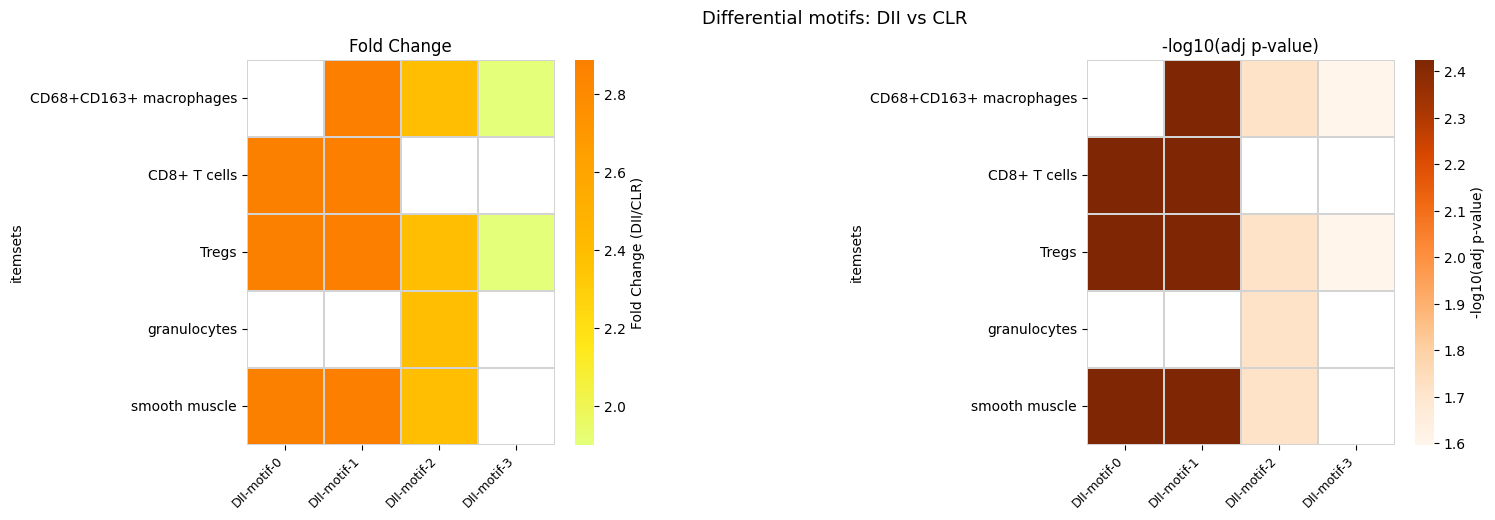
| 0 | (CD8+ T cells, Tregs, smooth muscle) | 0.050989 | 0.146879 | 0.003753 |
| 1 | (CD68+CD163+ macrophages, CD8+ T cells, Tregs,... | 0.048059 | 0.138878 | 0.003753 |
| 2 | (CD68+CD163+ macrophages, Tregs, granulocytes,... | 0.041341 | 0.098820 | 0.019282 |
| 3 | (CD68+CD163+ macrophages, Tregs) | 0.159009 | 0.302162 | 0.025271 |
Validate Differential Motifs with Enrichment Analysis
A motif being differential (more frequent in one condition) does not guarantee it is enriched (significantly over-represented compared to random expectation) in that condition. To confirm, run motif_enrichment_dist on the differential motifs within the condition where they are more frequent.
[12]:
for ds in [clr, dii]:
df = diff_motif[ds]
if len(df) == 0:
print(f"{ds}: No differential motifs to validate.\n")
continue
# Extract motif lists from the differential results
motif_lists = [list(m) for m in df["itemsets"]]
enrich_result = spm.motif_enrichment_dist(
ct=anchor_ct,
motifs=motif_lists,
dataset=ds,
max_dist=max_dist,
min_support=min_support,
)
n_enriched = enrich_result["if_significant"].sum()
print(f"{ds}: {len(df)} differential motifs → {n_enriched} also enriched")
display(enrich_result)
CLR: No differential motifs to validate.
DII: 4 differential motifs → 4 also enriched
| center | motifs | n_center_motif | n_center | n_motif | expectation | p-values | adj-pval | if_significant | |
|---|---|---|---|---|---|---|---|---|---|
| 0 | B cells | [CD8+ T cells, Tregs, smooth muscle] | 524 | 2774 | 14684 | 276.488664 | 4.919139e-47 | 1.967656e-46 | True |
| 1 | B cells | [CD68+CD163+ macrophages, CD8+ T cells, Tregs,... | 390 | 2774 | 13610 | 256.266053 | 4.003156e-17 | 8.006311e-17 | True |
| 2 | B cells | [CD68+CD163+ macrophages, Tregs] | 758 | 2774 | 33568 | 632.060167 | 7.534204e-09 | 1.004561e-08 | True |
| 3 | B cells | [CD68+CD163+ macrophages, Tregs, granulocytes,... | 269 | 2774 | 12513 | 235.610369 | 1.094891e-02 | 1.094891e-02 | True |
KNN-based Differential Motif Analysis
The KNN variant (differential_analysis_knn) defines neighborhoods by the k nearest neighbors rather than a fixed radius. This can provide complementary results, especially when cell density varies.
[13]:
k = 30
diff_motif_knn = spm.differential_analysis_knn(
ct=anchor_ct,
datasets=[clr, dii],
k=k,
min_support=min_support,
)
for ds in [clr, dii]:
print(f"KNN differential motifs in {ds}: {len(diff_motif_knn[ds])}")
KNN differential motifs in CLR: 0
KNN differential motifs in DII: 4
[14]:
diff_motif_knn[clr]
[14]:
| itemsets | support_CLR_mean | support_DII_mean | adj-pval |
|---|
[15]:
diff_motif_knn[dii]
[15]:
| itemsets | support_CLR_mean | support_DII_mean | adj-pval | |
|---|---|---|---|---|
| 0 | (CD68+CD163+ macrophages, Tregs, smooth muscle) | 0.058319 | 0.179174 | 0.023677 |
| 1 | (CD4+ T cells CD45RO+, CD68+CD163+ macrophages... | 0.107069 | 0.245905 | 0.023677 |
| 2 | (CD4+ T cells CD45RO+, CD68+CD163+ macrophages... | 0.032668 | 0.118155 | 0.023677 |
| 3 | (CD4+ T cells CD45RO+, CD68+CD163+ macrophages... | 0.093074 | 0.211532 | 0.030161 |
Validate KNN Differential Motifs with Enrichment Analysis
Same validation as above, but using motif_enrichment_knn to match the KNN neighborhood definition used in differential analysis.
[16]:
for ds in [clr, dii]:
df = diff_motif_knn[ds]
if len(df) == 0:
print(f"{ds}: No differential motifs to validate.\n")
continue
motif_lists = [list(m) for m in df["itemsets"]]
enrich_result = spm.motif_enrichment_knn(
ct=anchor_ct,
motifs=motif_lists,
dataset=ds,
k=k,
)
n_enriched = enrich_result["if_significant"].sum()
print(f"{ds}: {len(df)} differential motifs → {n_enriched} also enriched")
display(enrich_result)
CLR: No differential motifs to validate.
DII: 4 differential motifs → 4 also enriched
| center | motifs | n_center_motif | n_center | n_motif | expectation | p-values | adj-pval | if_significant | |
|---|---|---|---|---|---|---|---|---|---|
| 0 | B cells | [CD4+ T cells CD45RO+, CD68+CD163+ macrophages... | 535 | 2774 | 24457 | 460.506896 | 0.000075 | 0.000171 | True |
| 1 | B cells | [CD4+ T cells CD45RO+, CD68+CD163+ macrophages... | 256 | 2774 | 10846 | 204.222014 | 0.000102 | 0.000171 | True |
| 2 | B cells | [CD4+ T cells CD45RO+, CD68+CD163+ macrophages... | 438 | 2774 | 19761 | 372.084752 | 0.000128 | 0.000171 | True |
| 3 | B cells | [CD68+CD163+ macrophages, Tregs, smooth muscle] | 435 | 2774 | 20771 | 391.102292 | 0.007858 | 0.007858 | True |
Built-in Visualization
SpatialQuery provides plot_differential_pattern_heatmap for quick visualization of differential motif results.
[18]:
from SpatialQuery.plotting import plot_differential_pattern_heatmap
plot_differential_pattern_heatmap(diff_motif=diff_motif, ct=anchor_ct)
Custom Visualization
For more control over the layout, you can build custom heatmaps from the result DataFrame. Below we create side-by-side fold change and significance heatmaps.
[19]:
diff_motif[clr] = diff_motif[clr].copy()
diff_motif[dii] = diff_motif[dii].copy()
diff_motif[clr]["group"] = clr
diff_motif[dii]["group"] = dii
enrich = pd.concat([diff_motif[dii], diff_motif[clr]], axis=0)
enrich["fold_change"] = enrich[f"support_{dii}_mean"] / enrich[f"support_{clr}_mean"]
enrich["motif_group"] = enrich["group"] + "_motif_" + enrich.index.astype(str)
col_order = enrich["motif_group"].tolist()
enrich_expanded = enrich.explode("itemsets")
heatmap_fc = enrich_expanded.pivot_table(
index="itemsets", columns="motif_group", values="fold_change",
)[col_order]
heatmap_pval = enrich_expanded.pivot_table(
index="itemsets", columns="motif_group", values="adj-pval",
)
heatmap_pval = -np.log10(heatmap_pval)[col_order]
col_labels = [c.replace("_", "-") for c in col_order]
heatmap_fc.columns = col_labels
heatmap_pval.columns = col_labels
fig, (ax1, ax2) = plt.subplots(1, 2, figsize=(16, 5), gridspec_kw={"wspace": 1.1})
sns.heatmap(
heatmap_fc, cmap="Wistia", linewidths=0.1, linecolor="lightgrey",
square=True, cbar_kws={"label": "Fold Change (DII/CLR)"}, ax=ax1,
)
ax1.set_title("Fold Change")
ax1.set_xlabel("")
ax1.set_xticklabels(ax1.get_xticklabels(), rotation=45, ha="right", fontsize=9)
sns.heatmap(
heatmap_pval, cmap="Oranges", linewidths=0.1, linecolor="lightgrey",
square=True, cbar_kws={"label": "-log10(adj p-value)"}, ax=ax2,
)
ax2.set_title("-log10(adj p-value)")
ax2.set_xlabel("")
ax2.set_xticklabels(ax2.get_xticklabels(), rotation=45, ha="right", fontsize=9)
fig.suptitle(f"Differential motifs: {dii} vs {clr}", fontsize=13)
plt.tight_layout()
plt.show()
[ ]: