Single-dataset: Differential Expression Analysis
This example demonstrates how to identify differentially expressed genes between motif-positive and motif-negative anchor cells within a single spatial dataset.
After identifying an enriched motif, SpatialQuery can split anchor cells into two groups: those surrounded by the motif (motif+) and those not (motif−). Comparing gene expression between these groups reveals genes whose expression is associated with specific spatial contexts.
Dataset: seqFISH mouse organogenesis E8.5 embryo (17,806 cells × 351 genes) Key API: spatial_query.de_genes
Setup
[2]:
import warnings
warnings.filterwarnings("ignore")
import anndata as ad
import numpy as np
import pandas as pd
from SpatialQuery import spatial_query
Load Data & Initialize
[4]:
DATA_DIR = "../data/mouse_organogenesis"
adata = ad.read_h5ad(f"{DATA_DIR}/embryo1.h5ad")
adata
[4]:
AnnData object with n_obs × n_vars = 17806 × 351
obs: 'embryo', 'pos', 'z', 'x_global', 'y_global', 'x_global_affine', 'y_global_affine', 'embryo_pos', 'embryo_pos_z', 'Area', 'UMAP1', 'UMAP2', 'celltype_mapped_refined', 'segmentation_vertices_x_global_affine', 'segmentation_vertices_y_global_affine'
var: 'gene'
uns: 'celltype_mapped_refined_colors'
obsm: 'X_spatial', 'X_umap'
Inspect adata.obsm for spatial coordinates, adata.obs for cell type labels, and adata.var for gene names. Then set the corresponding column names below.
[5]:
spatial_key = "X_spatial"
label_key = "celltype_mapped_refined"
feature_name = "gene"
dataset_name = "mouse_embryo1"
[6]:
sp = spatial_query(
adata=adata,
dataset=dataset_name,
spatial_key=spatial_key,
label_key=label_key,
feature_name=feature_name,
build_gene_index=False,
if_lognorm=True,
if_normalize_spatial_coord=True,
)
Auto-normalizing spatial coordinates: mean nearest neighbor distance = 1.0
Scale factor: 51.7200
build_gene_index is False. Using adata.X for gene expression analysis.
Log normalizing the expression data... If data is already log normalized, please set if_lognorm to False.
Define Anchor Cell Type and Motif
We use Gut tube as the anchor and test the motif [Splanchnic mesoderm, Endothelium]. First, test the enrichment and retrieve the cell IDs of motif-positive anchor cells using motif_enrichment_dist with return_cellID=True.
Note: In the single-dataset API,
return_cellID=Trueaddscenter_idandneighbor_idcolumns directly to the returned DataFrame. The multi-FOV version returns a 3-tuple(DataFrame, motif_id_dict, center_id_dict)instead.
[7]:
anchor_ct = "Gut tube"
motif = ["Splanchnic mesoderm", "Endothelium"]
max_dist = 8
motif_result = sp.motif_enrichment_dist(
ct=anchor_ct,
motifs=motif,
max_dist=max_dist,
return_cellID=True,
)
motif_result
[7]:
| center | motifs | n_center_motif | n_center | n_motif | expectation | p-values | neighbor_id | center_id | if_significant | |
|---|---|---|---|---|---|---|---|---|---|---|
| 0 | Gut tube | [Endothelium, Splanchnic mesoderm] | 322 | 1464 | 3117 | 256.278109 | 0.000002 | [6151, 6163, 6168, 6182, 6188, 14381, 6199, 62... | [529, 536, 537, 547, 550, 4493, 4611, 4613, 46... | True |
Alternative: KNN-based Cell ID Retrieval
You can also retrieve motif-positive cell IDs using the KNN neighborhood definition instead of a fixed radius.
[8]:
k = 50
motif_result_knn = sp.motif_enrichment_knn(
ct=anchor_ct,
motifs=motif,
k=k,
return_cellID=True,
)
motif_result_knn
[8]:
| center | motifs | n_center_motif | n_center | n_motif | expectation | p-values | neighbor_id | center_id | if_significant | |
|---|---|---|---|---|---|---|---|---|---|---|
| 0 | Gut tube | [Endothelium, Splanchnic mesoderm] | 289 | 1464 | 2845 | 233.914411 | 0.000027 | [4471, 4482, 4497, 4510, 4516, 4526, 4535, 453... | [4493, 4619, 4621, 4623, 4654, 4656, 4662, 466... | True |
Split Anchor Cells into Motif+ and Motif− Groups
The center_id column contains the indices of motif-positive anchor cells. We derive the motif-negative group as the complement.
[9]:
center_id = motif_result["center_id"].iloc[0]
all_center_id = np.where(sp.labels == anchor_ct)[0]
non_center_id = np.setdiff1d(all_center_id, center_id)
print(f"Motif+ anchor cells: {len(center_id)}")
print(f"Motif− anchor cells: {len(non_center_id)}")
Motif+ anchor cells: 322
Motif− anchor cells: 1142
Run Differential Expression Analysis
de_genes performs a statistical test (t-test, Wilcoxon, or Fisher’s exact) to identify genes differentially expressed between two groups of cells.
Key parameters:
Parameter |
Description |
|---|---|
|
Cell indices for group 1 (motif+) |
|
Cell indices for group 2 (motif−) |
|
Statistical test: |
|
Minimum fraction of cells expressing a gene (in either group) to include it |
|
Significance threshold for adjusted p-values |
Output columns:
Column |
Description |
|---|---|
|
Gene name |
|
Fraction of cells expressing the gene in group 1/2 |
|
Absolute difference in proportions |
|
Log2 fold change (group 1 vs group 2) |
|
Raw p-value |
|
FDR-adjusted p-value |
|
Which group the gene is upregulated in ( |
[10]:
de_result = sp.de_genes(
ind_group1=center_id,
ind_group2=non_center_id,
min_fraction=0.05,
method="t-test",
alpha=0.05,
)
print(f"Number of DE genes: {len(de_result)}")
de_result.head(10)
Testing 351 genes ...
Number of DE genes: 80
[10]:
| gene | proportion_1 | proportion_2 | abs_difference | log2fc | p_value | adj-pval | de_in | |
|---|---|---|---|---|---|---|---|---|
| 0 | Dkk1 | 0.111801 | 0.243433 | 0.131631 | -1.586187 | 8.074493e-14 | 1.778115e-11 | group2 |
| 1 | Wnt5a | 0.319876 | 0.472855 | 0.152979 | -1.045780 | 1.013171e-13 | 1.778115e-11 | group2 |
| 2 | Osr1 | 0.652174 | 0.464974 | 0.187200 | 0.943958 | 2.715462e-12 | 3.177090e-10 | group1 |
| 3 | T | 0.161491 | 0.260946 | 0.099455 | -1.527118 | 6.675586e-12 | 5.857827e-10 | group2 |
| 4 | Meis1 | 0.503106 | 0.345009 | 0.158097 | 0.959926 | 1.492688e-09 | 1.047867e-07 | group1 |
| 5 | Otx2 | 0.180124 | 0.271454 | 0.091329 | -1.065929 | 1.480319e-08 | 8.659869e-07 | group2 |
| 6 | Hoxb1 | 0.394410 | 0.270578 | 0.123832 | 1.148066 | 1.902122e-08 | 9.537782e-07 | group1 |
| 7 | Pitx1 | 0.173913 | 0.251313 | 0.077400 | -1.158265 | 2.657183e-08 | 1.165839e-06 | group2 |
| 8 | Gata5 | 0.413043 | 0.280210 | 0.132833 | 1.014293 | 4.837701e-08 | 1.886703e-06 | group1 |
| 9 | Cdh2 | 0.267081 | 0.378284 | 0.111203 | -0.793237 | 8.300411e-07 | 2.703245e-05 | group2 |
Visualize Top DE Genes
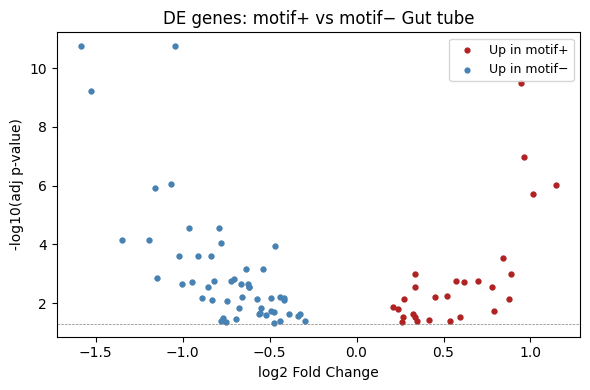
A volcano plot provides a quick overview of effect size (log2 fold change) vs significance.
[11]:
import matplotlib.pyplot as plt
fig, ax = plt.subplots(figsize=(6, 4))
de = de_result.copy()
de["-log10(adj-pval)"] = -np.log10(de["adj-pval"].clip(lower=1e-300))
ax.scatter(
de["log2fc"], de["-log10(adj-pval)"],
c="grey", s=10, alpha=0.5,
)
# Highlight significant genes
sig = de[de["adj-pval"] < 0.05]
up = sig[sig["de_in"] == "group1"] # motif+
down = sig[sig["de_in"] == "group2"] # motif-
ax.scatter(up["log2fc"], up["-log10(adj-pval)"], c="firebrick", s=12, label="Up in motif+")
ax.scatter(down["log2fc"], down["-log10(adj-pval)"], c="steelblue", s=12, label="Up in motif−")
ax.axhline(-np.log10(0.05), color="grey", linestyle="--", linewidth=0.5)
ax.set_xlabel("log2 Fold Change")
ax.set_ylabel("-log10(adj p-value)")
ax.set_title(f"DE genes: motif+ vs motif− {anchor_ct}")
ax.legend(fontsize=9)
plt.tight_layout()
plt.show()